PROTAC偶联热潮来袭!一文读懂DAC设计要点、代表性案例
转自 肖恩 医药魔方Pro 2022-06-05 21:00 发表于江苏
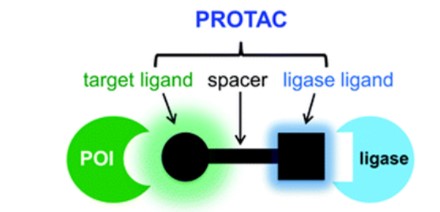
蛋白水解靶向嵌合体(PROTAC)正为生物学与药物化学领域带来革命性的变化。PROTAC由靶蛋白配体、E3连接酶配体与间隔基团[区别于抗体偶联降解剂中的连接基团,这里特地使用“间隔基团”(spacer)而不是PROTAC文献中常见的“连接基团”(linker)]组成。
PROTAC结构组成(来源:ChemicalSociety Reviews)
由于抗体偶联药物(ADC)不断显现临床可行性与商业价值,近年来有不少研究小组试图通过仿照ADC的设计原理,将PROTAC与单抗(mAb)连接,提高PROTAC体内递送效率。这种叫做抗体偶联降解剂(Degrader-antibody conjugates,DAC)的药物形式可克服未偶联PROTAC分子的以下问题:(1)由于理化性质与DMPK性质(尤其E3连接酶未选用CRBN)导致体内递送效率低;(2)为实现充分的暴露量而采用的复杂的处方;(3)PROTAC非特异性靶向。
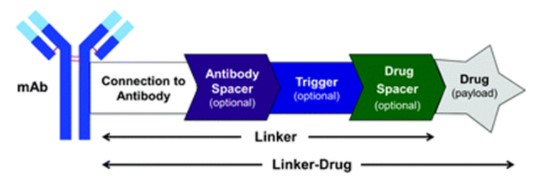
ADC通过连接基团将细胞毒性小分子药物(有效载荷)与mAb结合。为提高ADC体内活性,有时候结构中还会有(1)一个将连接基团与抗体隔开的间隔元件(Antibody Spacer);(2)激活细胞内裂解的化学或生物触发元件(Trigger);(3)将触发元件与小分子药物隔开的第二个间隔元件(Drug Spacer)。(备注:下图中不同色块对应于后文中插图的对应模块)
ADC结构组成(来源:ChemicalSociety Reviews)
通过化学合成将小分子药物与连接基团缀合,随后通过偶联技术结合到抗体表面。连接到抗体表面的药物无法与靶标结合。ADC结构中的抗体与细胞表面抗原结合后内化整个ADC分子。ADC靶向的抗原必须在靶组织具有较高的表达量,能迅速内化ADC并具有适当的转运能力,而在非靶组织中表达量相对较低。
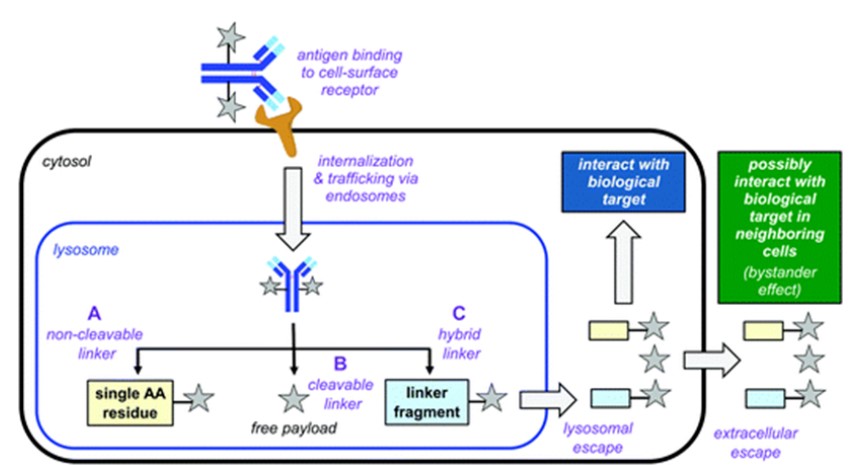
内化后的ADC分子随后转运到细胞溶酶体中释放小分子药物。目前已开发出多种溶酶体响应的裂解方式,如酸水解、二硫键还原、多肽酶切。具有不同裂解方式的ADC在体循环中可保持稳定,仅在靶细胞中的溶酶体释放小分子。小分子通过主动转运移出溶酶体,扩散到细胞质中,随后与靶标相互作用。这就要求小分子药物在溶酶体中必须非常稳定,才能顺利与靶标相互作用。
ADC作用机制。(A)无可裂解连接基团:抗体被溶酶体酶代谢,药物与连接基团和一小部分抗体结构(通常是单个氨基酸)结合;(B)含可裂解基团:触发元件在溶酶体中激活断裂,释放游离小分子;(C)部分情况下断裂后小分子会与连接基团片段结合(来源:Chemical Society Reviews)
有些情况下,释放的小分子药物可能进入临近的细胞,这些细胞可能表达、也可能未表达靶抗原。这种“旁观者效应”可以杀伤不表达靶抗原的相关细胞(缓解肿瘤异质性问题),但也可能对非靶细胞造成脱靶毒性。
目前,ADC药物的偶联技术已经有了相当大的发展。第一类技术是使用N-羟基琥珀酰亚胺(NHS)酯的活化羧基与抗体的赖氨酸残基进行偶联,首个批准的ADC药物Mylotarg是即采用这种技术实现的,但这种方法产生的药物抗体比(DAR,即单个mAb上偶联的药物个数)变化范围较宽,在2~7个。第二类技术是通过使用巯基特异性试剂(如马来酰亚胺)与半胱氨酸反应来合成的。利用马来酰亚胺法偶联可形成DAR为4(部分还原,均一度较差)或8(完全还原,均一度强)的ADC分子。
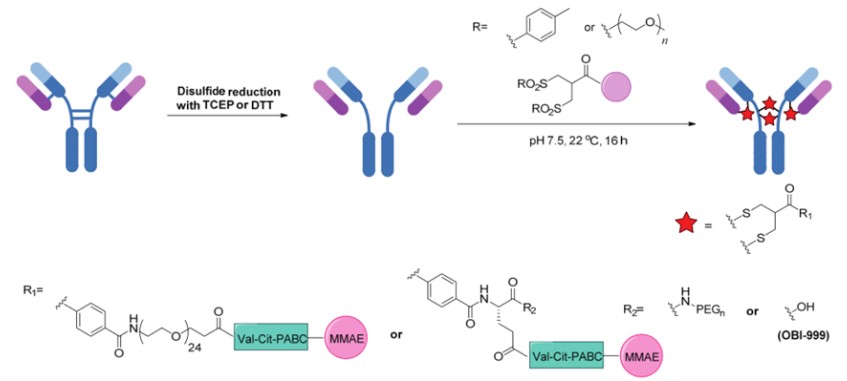
二硫键桥连法是通过对抗体中的4对链间二硫键,用还原性化合物[如三(2羧乙基)膦 (TCEP) ]进行还原,然后利用与半胱氨酸选择性反应的试剂对抗体二硫键进行重新桥连的同时引入细胞毒性药物。该方法不仅能够保持原有二硫键的稳定作用,并且可以控制每对二硫键只偶联一个连接分子,从而实现定位偶联。
使用双砜化合物重新桥连抗体的链间二硫键产生均一的ADC(来源:《自然杂志》)
DAR的数量要求与小分子药物活性有关,小分子活性越强,需要的DAR数量越低。某些ADC中的小分子药物抑制微管蛋白聚合破坏微管活性,或通过烷基化直接杀伤DNA,也可抑制DNA拓扑异构酶1活性杀伤肿瘤。常见的ADC的小分子在体外都具有强大的细胞毒性特征,抗增殖IC50可达亚纳摩尔级,DAR仅需2-4即可发挥作用。而DNA拓扑异构酶1抑制剂活性稍低,有效载荷为DNA拓扑异构酶1抑制剂的ADC中,DAR需达到4-8才足以触发充分的细胞杀伤作用。但随着DAR升高, ADC会有聚集倾向,不利于发挥肿瘤杀伤作用。
在开发DAC时,由于PROTAC分子的特殊性,不能简单照搬ADC的递送策略。不同于ADC中使用的广谱细胞毒性小分子药物,DAC中的PROTAC通常只对特定的肿瘤和/或组织或细胞具有靶向活性。因此,抗原的选择不仅需要满足内化DAC并运输到溶酶体中的功能,还需要在PROTAC靶组织(或肿瘤、细胞)上高表达。这种抗原在其他组织或细胞上可能也会有较低水平的表达,只要这类组织或细胞对该PROTAC具有良好的耐受性即可避免脱靶毒性。
由于PROTAC体外活性很多情况下不如小分子药物,因此需要提高DAC中的DAR以发挥药效(即DAC>4)。但是,提高DAR可能导致DAC聚集,对体内药动学(PK)造成不良影响。然而,相较于小分子药物,PROTAC体积更大,亲脂性更强,这种差异会使聚集和PK问题更加严重,需要开发新的连接基团和偶联方法解决上述问题。
许多PROTAC没有可以用于与可裂解连接基团共价结合的位点(氨基)。因此,需要考虑是否需要对PROTAC结构修饰,引入活性位点(但有可能改变PROTAC生理活性);或利用PROTAC现有官能团(如羟基、酚羟基)并开发新的偶联技术。而开发不可裂解基团也需要考虑同样的问题,而溶酶体降解后仍附着在PROTAC分子上的连接基团不得干扰其生物活性。
此外,DAC开发中其他需要解决的问题还包括:(1)PROTAC在溶酶体中的稳定性;(2)PROTAC溶酶体逃逸功能;(3)PROTAC旁观者效应。后两者受到PROTAC细胞通透性的影响,仍是这一领域当下研究的重点。受细胞通透性影响无法发挥生物学活性的PROTAC可利用DAC中的mAb进入细胞发挥作用,但这类PROTAC旁观者效应预计较小。这种情况下,需要使用不依赖细胞的方法评估该PROTAC生成三元复合物的性能。
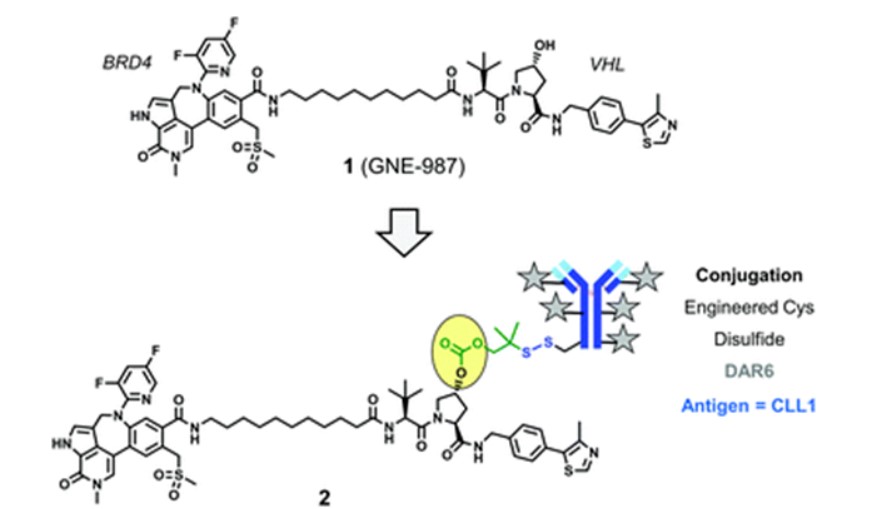
GNE-987(化合物1)是一种降解溴结构域蛋白4(BRD4)的PROTAC,由VHL配体、BRD4配体与间隔基团组成。通过一种含有二硫键的可裂解连接基团将GNE-987偶联到靶向C-型凝集素样分子1(CLL1)的抗体上构成了DAC(偶联物2)。这种DAC利用VHL配体结构中羟脯氨酸的羟基与连接基团结合。单个抗体的重链与轻链共可以通过二硫键结合6个PROTAC分子。
偶联物2(来源:ChemicalSociety Reviews)
单次静脉注射后,偶联物2药物在急性髓系白血病(AML)HL-60与EOL-1异种移植模型中显示了强大、剂量依赖的体内活性。而未偶联的化合物1的活性受不利的PK性质干扰,体内活性较差。上述结果表明,DAC可以克服PROTAC不理想的PK特性,实现可接受的PROTAC溶酶体稳定性及适当的抗原靶向能力。
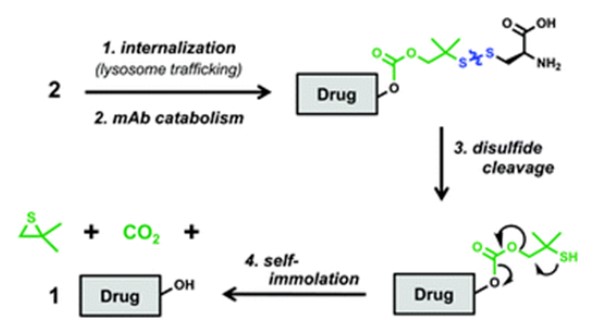
化合物2的抗体区域与肿瘤细胞表面CLL1受体结合,偶联物随后内化并运输到溶酶体。抗体在溶酶体中水解为氨基酸,但仍有一个半胱氨酸残基通过二硫键与连接基团结合。二硫键随后被还原并发生重排,暴露VHL配体中羟基,释放游离GNE-987。
偶联物2释放游离PROTAC的机理(来源:Chemical Society Reviews)
在化合物2公布不久后,又出现了第二个靶向BDR4的DAC药物。在这个药物中,利用mAb链间半胱氨酸残基,通过二硫键桥连法技术将4个PROTAC分子(化合物3)结合到抗体表面。在偶联过程中,利用VHL配体结构中的羟基与一个非裂解基团形成酯键,最后再利用点击化学中的环张力促进的叠氮-炔环加成(SPAAC)形成环状炔烃,构成完整的DAC药物(偶联物4)。细胞摄取后的化合物4药物在溶酶体中完成酯键水解,释放游离的PROTAC分子。

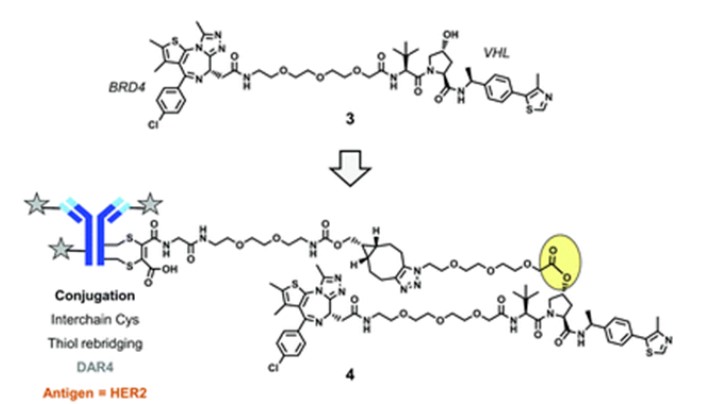
偶联物4(来源:ChemicalSociety Reviews)
偶联物4靶向人类表皮生长因子受体2(HER2),这是一种在在ADC中应用广泛的抗原。体外实验表明,偶联物4在HER2阳性细胞中以剂量依赖的方式降解了BRD4,而在HER2阴性细胞中未观察到上述现象。
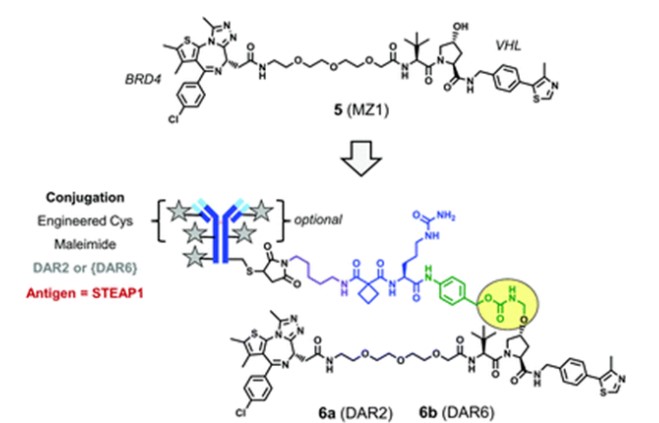
将降解BRD4的PROTAC药物MZ1(化合物5)通过新型连接基团与抗体结合形成偶联物6,该接连基团中含有酶裂解拟肽触发元件和一个药物间隔元件。偶联物6的抗体识别前列腺六段跨膜上皮抗原1(STEAP1),该抗原在前列腺表面高度表达。
偶联物6(来源:ChemicalSociety Reviews)
诱导表达STEAP1抗原的PC3前列腺癌细胞(PC3-S1)中,DAR为6的偶联物6b比DAR为2的偶联物6a降解活性更强。因此,提高DAR可以改善偶联物的生物活性。然而,制备更高DAR偶联物需要研发人员丰富的经验,药物分子也受多种因素影响(mAb抗原、连接基团结构、PROTAC亲脂性等)。
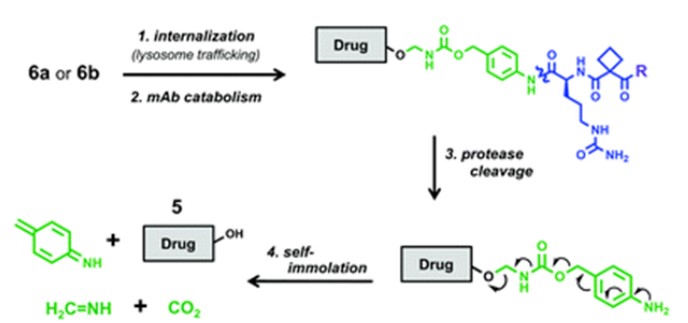
偶联物6的抗体与STEAP1受体结合后,药物被内化、转运到细胞溶酶体,经历抗体水解、拟肽断裂,药物与间隔元件发生重排,释放游离的PROTAC分子。
偶联物6裂解途径(来源:ChemicalSociety Reviews)
由于PROTAC药物结构的特殊性,有时需要向PROTAC结构中引入胺或苯胺构成连接基团的结合位点。例如,通过向化合物5结构的不同位置中引入伯胺、仲胺或苯胺,修饰后的PROTAC分子通过氨基甲酸酯结合到酶裂解拟肽连接基团,随后与靶向STEAP1抗体偶联。
PC-S1细胞实验发现,与化合物5相比,修饰后PROTAC分子对BRD4的降解能力下降,可能是引入极性官能团后破坏了PROTAC的细胞膜通透性。细胞外表面等离子体共振(SPR)实验表明,修饰后的PROTAC分子可与BRD4与VHL形成三元复合物,三元复合物的半衰期与BRD4降解活性之间存在较强的相关性。
基于对化合物5结构改造设计的DAC(来源:Chemical Society Reviews)
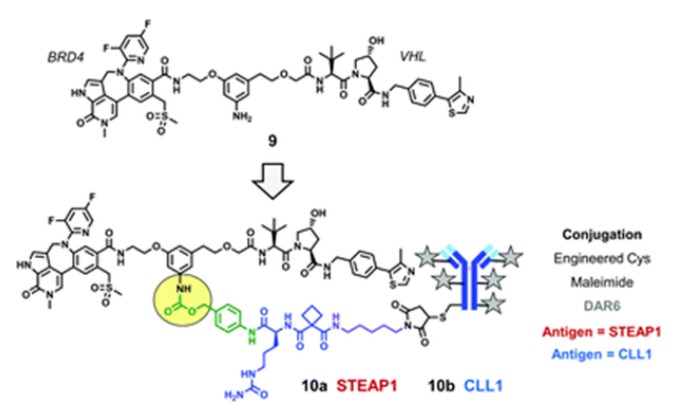
为进一步提高BRD4的降解活性,获得更强的抗细胞增殖活性,基于亲和力更强的BRD4配体开发了一系列新的BRD4降解剂(化合物9)。将化合物9与靶向STEAP1的mAb偶联,形成了多种具有体外抗增殖活性的DAC。
例如,将化合物9利用拟肽连接基团与氨基甲酸酯分别结合到靶向STEAP1或CLL1的mAb上,分别构成偶联物10a和偶联物10b。DAC 10a在PC3-S1细胞中具有明显的抗原依赖性抗增殖作用;DAC 10b在AML HL-60异种移植模型展现了抗原依赖的肿瘤杀伤作用。
偶联物10(来源:ChemicalSociety Reviews)
然而,将偶联物2的抗体替换为识别STEAP1的mAb,形成DAR为6的偶联物,将导致DAC聚集而丧失生物活性。因此,这些结果表明,利用高亲脂性PROTAC开发高DAR偶联物时必须同时考虑mAb抗原以及连接基团-药物,才能开发具有活性的DAC药物。
偶联后的10a、10b对巨核细胞的毒性比未偶联化合物9降低,这表明通过将PROTAC与mAb偶联可以提高DAC的临床前治疗指数,但具体的结论仍需通过许多额外的实验证明。
最近报道了一种新的降解BRD4的DAC药物,这种DAC是基于CRBN设计的。降解BRD4的PROTAC(化合物11)通过链间半胱氨酸残基与靶向HER2 mAb结合,形成偶联物12。细胞实验中,偶联物12可以降解HER2阳性细胞BT-474中的BRD4,但不能降解HER2阴性细胞MDA-MB-231。
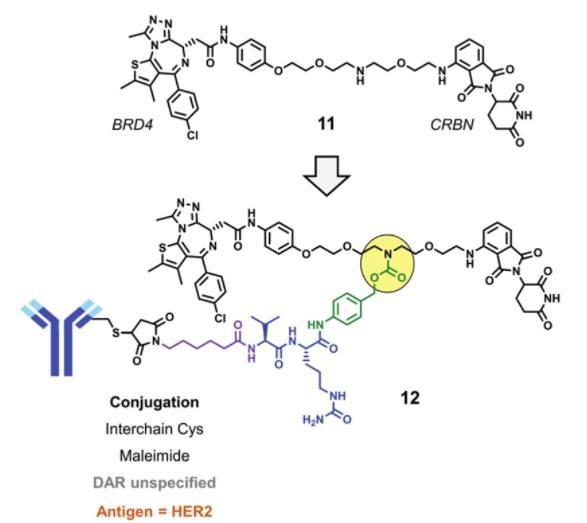
偶联物12(来源:ChemicalSociety Reviews)
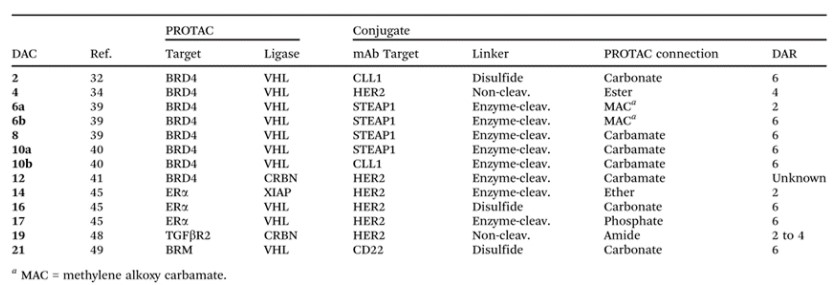
目前,DAC仍处于起步阶段,但已经发现了多个具有体外和/或体内生物活性的候选药物。这些DAC结构中的PROTAC分子利用了不同的E3连接酶,靶向不同的目标蛋白。此外,多种新型连接基团以及抗体偶联技术被应用于DAC中。
部分DAC药物信息(来源:ChemicalSociety Reviews)
绝大多数DAC分子的DAR(6)比主流ADC(2-4)更大,但这种增加对DAC开发是否为通用规则还需继续研究。多个研究表明,DAC的活性有赖于细胞表面抗原,表明DAC药物能够将对应的PROTAC递送到特定肿瘤和/或细胞中。
基于上述结果,DAC模式的前景光明。但是以下问题仍需要解决:(1)哪些PROTAC分子适合开发成DAC;(2)何种偶联方式能最好得保留(甚至增强)PROTAC分子的生物学活性。
参考资料:
[1] Peter S. Dragovich. Degrader-antibody conjugates. Chemical Society Reviews. 2022.
[2] 黄容 et al. 基于二硫键桥连构建均一性抗体. 自然杂志. 2021.