详细内容
转自:同写意 2022-06-05 19:08 发表于江苏以下文章来源于Genhouse ,作者Jie Jack Li 团队

每棵巨大的苹果树都是从一粒小小的苹果种子开始的。用于拯救生命的药物的发现则是从苗头化合物(Hits)或者先导化合物(Leads)开始的。
在小分子药物开发项目中的药物化学部分,我们最开始通常需要一个化学结构作为起点,也就是苗头化合物或者先导化合物。那么它们究竟是从哪里来的呢?它们的主要来源包括(i)非理性的药物设计(偶然发现);(ii)天然产物;(iii)高通量筛选(HTS);(iv)基于片段的药物发现(FBDD);(v)DNA-编码化合物库(DEL);等。
对于今天的药物化学家来说,这是一个激动人心的时代。许多技术的进步使我们能够更容易地发现苗头化合物,尤其是对于那些非常有挑战性的“不可成药”靶点。本文简要介绍一种相对较新但非常有效的药物发现策略:基于片段的药物发现(FBDD),又称为基于骨架的药物设计。
截止到2021年底已经有六个基于片段衍生的药物被美国FDA批准上市,印证了FBDD在药物发现方面的威力(图1)。这六个药物分别是Plexxikon公司的B-RafV660E抑制剂vemurafenib(1)和CSF1R & KIT抑制剂pexidartinib(2);艾伯维公司的Bcl-2抑制剂venetoclax(3);Astex公司的FGFR3抑制剂erdafitinib(4);安进公司的KRASG12C抑制剂sotorasib(5)以及诺华公司的Bcr-Abl-1变构抑制剂asciminib(6)。值得注意的是,除了艾伯维公司的Bcl-2抑制剂venetoclax(3)是以蛋白–蛋白相互作用作为靶点外,市场上其他五个药物都是以蛋白激酶作为靶点FBDD的确在蛋白激酶抑制剂的发现上富有成效。【1】

图1:基于片段衍生批准上市的药物
已经有数十种基于片段衍生的药物进入II期临床试验,还有更多的处在I期临床试验阶段。
介绍
与高通量筛选(HTS)的化合物库集不同,片段化合物库通常不是很大,数量从几十到数千不等。顾名思义片段通常是重原子数小于20的小分子(分子量在100–250之间)。片段的官能团数量较少,刚好大到能与目标蛋白形成足够的相互作用,又小到将不利的相互作用降到最低。通常片段与靶标的亲和力相对较弱,需要较高的暴露浓度(微摩尔至毫摩尔级别),因此检测片段与靶标片段的结合需要灵敏度较高的筛选方法——核磁共振(NMR)和表面等离子体共振(SPR)技术是检测直接结合的常用方法,此外通过浸泡方式的蛋白–配体X-射线晶体衍射方法在片段的发现中也有许多应用。由于这些生物物理方法只能检测结合,因此还需要一个正交试验来去除非特异性结合的片段。
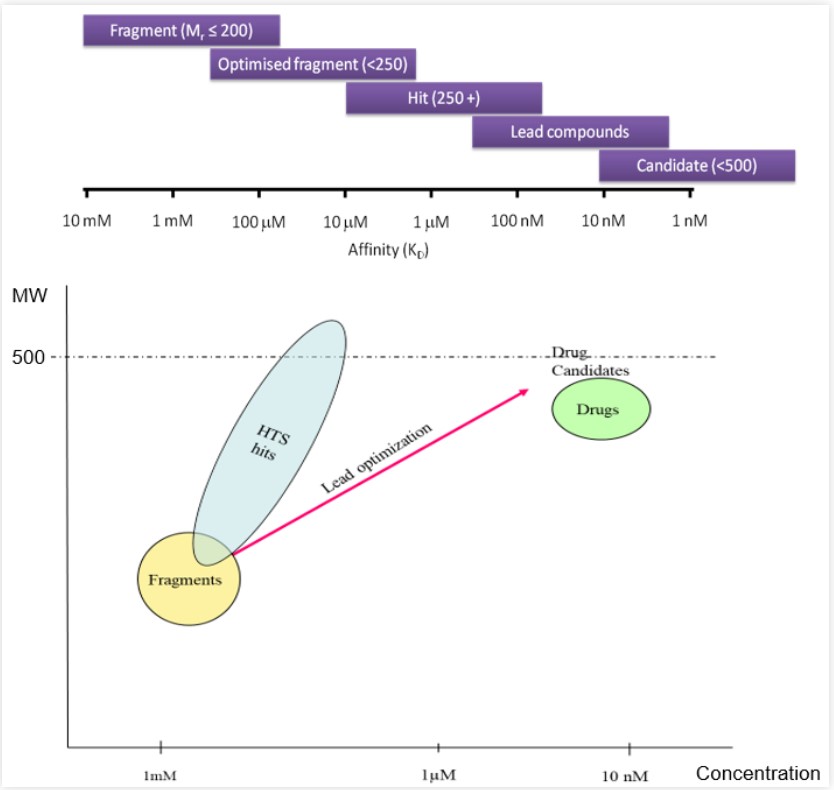
图2:片段的特征
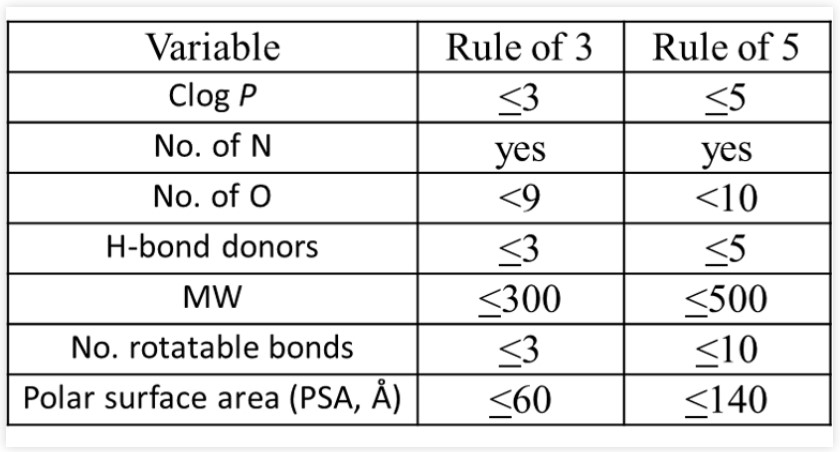
Table 1. RO3 and RO5
X-Ray晶体学指导的FBDD
Plexxikon公司的vemurafenib(1)是第一个在共晶学指导下通过FBDD策略开发并上市的药物。2002年,B-RafV660E的等位突变逐渐被发现可以作为癌症治疗的靶点。由于B-RafV660E是当时已知最常见的致癌蛋白激酶突变,而且只存在于依赖RAF/MEK/ERK通路的肿瘤中,Plexxikon公司迅速跟进,开始针对这一靶点开发药物。以200 mM的化合物浓度高通量筛选一个由20000个片段化合物组成的化合物库(分子量介于150-350之间,氢键的供体和受体 < 8,可旋转键数较少),最终得到238个苗头化合物。7-氮杂吲哚(7)是其中之一,与PIM1激酶的共晶结构显示,它结合在该蛋白的ATP结合位点。
同时,共晶显示3-苯胺-7-氮杂吲哚(8)同样可以与PIM1激酶结合,IC50大约为100 mM。化合物8的7-氮杂吲哚骨架代表了一种可以与激酶铰链区结合的通用框架片段,这一骨架通常可以与激酶的铰链区形成两个关键的氢键相互作用。通过一些微小的改动进一步得到了苄基-7-氮杂吲哚(9),共晶显示9可以与另一种蛋白FGFR1结合,并且对FGFR1的IC50为1.9 mM。
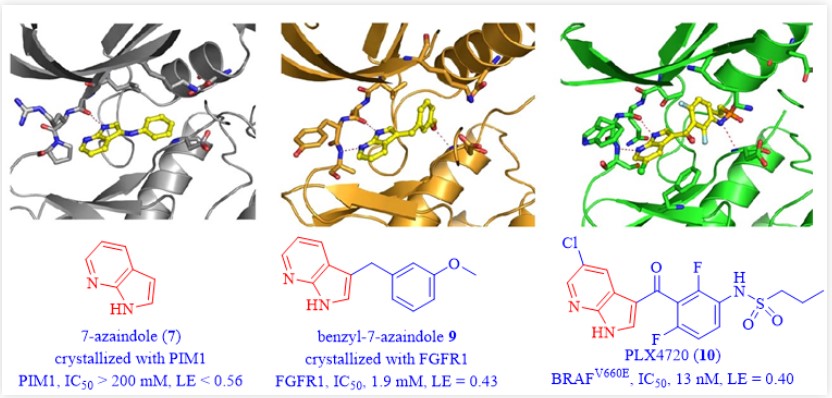
图3:PLX4720(10)的发现
进一步的构效关系研究得到了PLX4720(10)【4】,是一个高活性、高选择性(包括野生型B-Raf和很多其他激酶)的B-RafV660E抑制剂,IC50为13 nM(图3)。通过在10的7-氮杂吲哚母核上的5-位氯原子位置引入氯苯基就得到了vemurafenib(1)【5】。vemurafenib(1)对BRAF(31 nM)与c-RAF-1(48 nM)表现出类似的抑制活性,并且对其他激酶也有一定的选择性,包括野生型B-Raf(100 nM)。之所以选择vemurafenib(1)而不是10进一步开发,是因为它在比格犬和食蟹猴中的药代动力学性质更优。于是FDA于2011年批准vemurafenib(1)上市,用于BRAF突变的转移性黑色素瘤的治疗。【6】
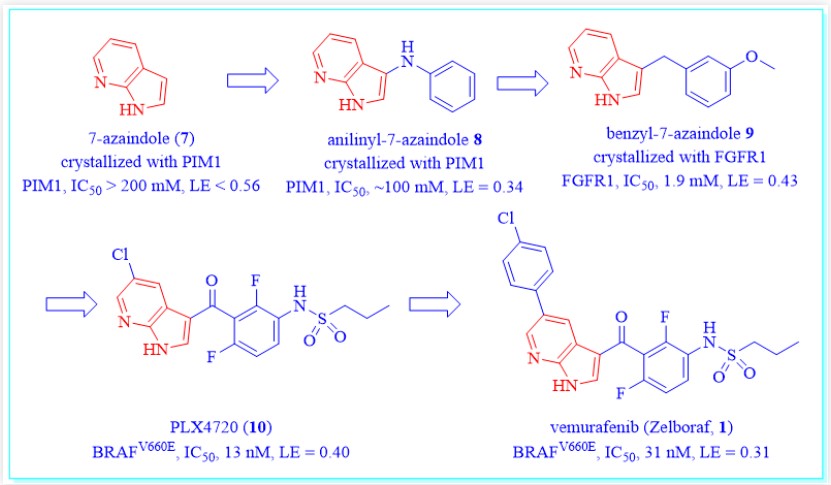
图4:vemurafenib(1)的发现过程
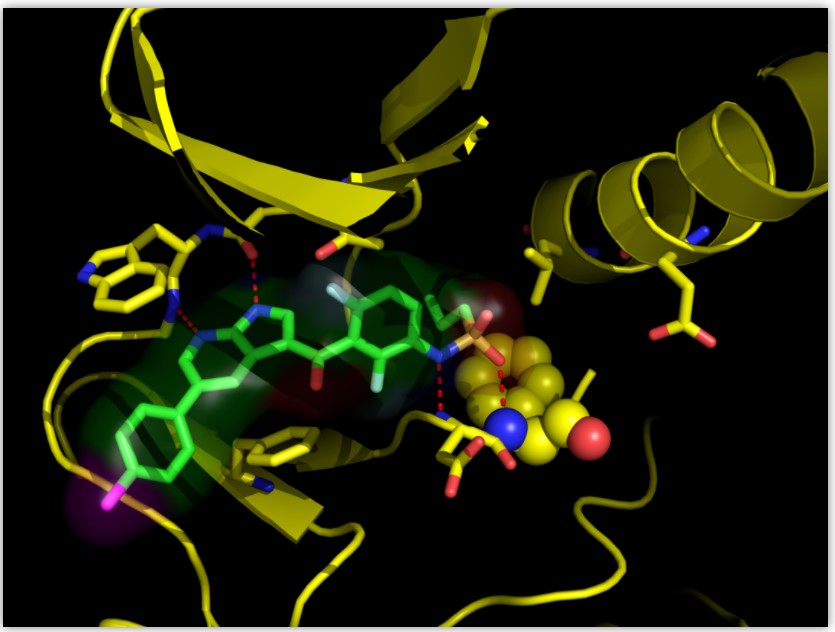
图5:vemurafenib(1)与B-RafV660E的结合模式
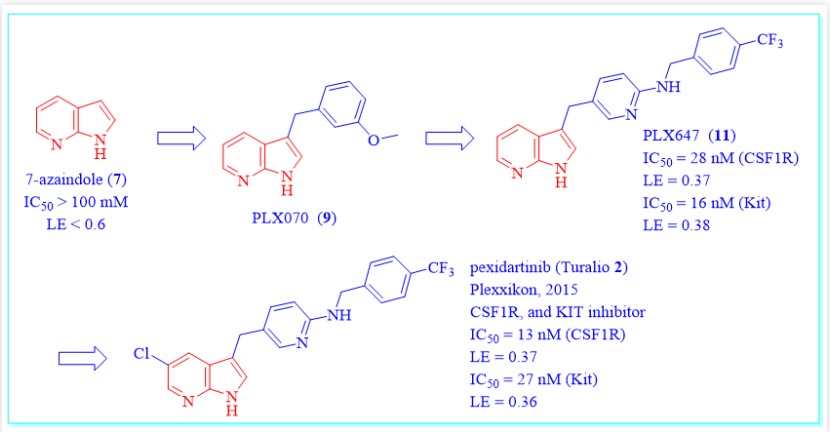
图6:pexidartinib(2)的发现过程
X-Ray NMR指导的FBDD
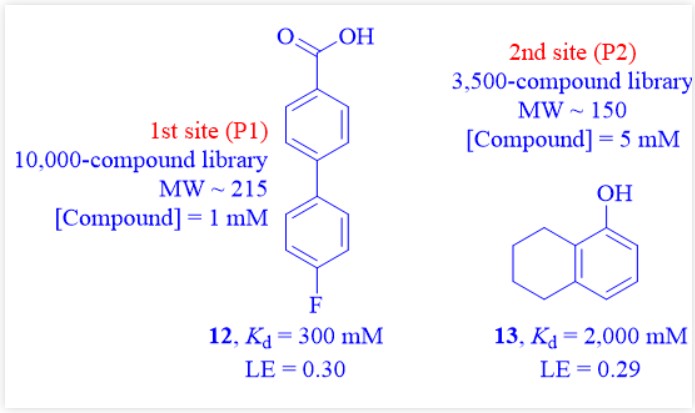
图7:片段12和13的结构
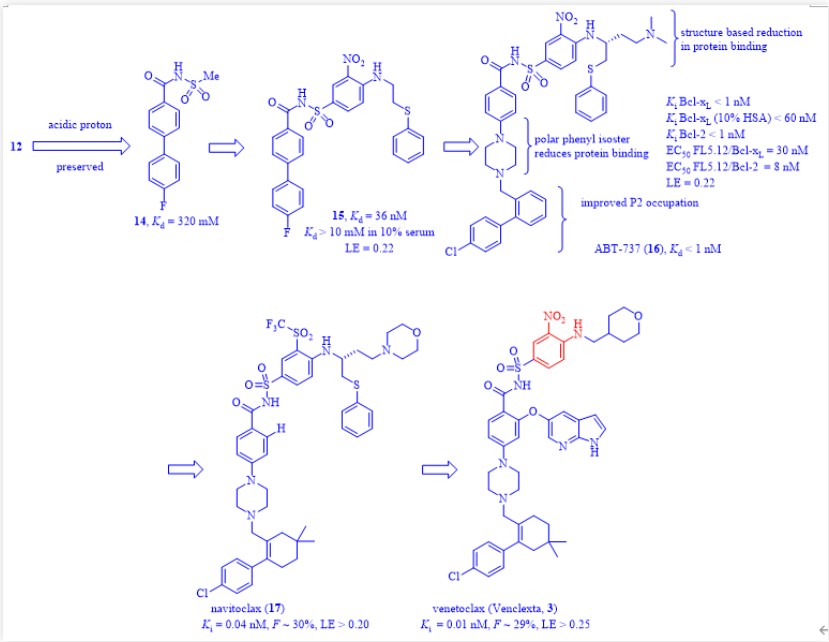
图8:venetoclax(3)的发现
片段结合的筛选还有许多其他的方法,例如表面等离子体共振(SPR),虚拟对接筛选(virtual docking screen)等,(图9)之后有机会可以单独些写一篇文章讨论。
更令人兴奋的是,现在已经建立了很多专门的片段库,用于发现表观遗传学靶点的苗头化合物。【12】 FBDD在RNA靶点的应用上也越来越受欢迎,【13】并且基于片段的共价配体的发现也已经在共价抑制剂的研发过程中做出了重要的贡献。【14】
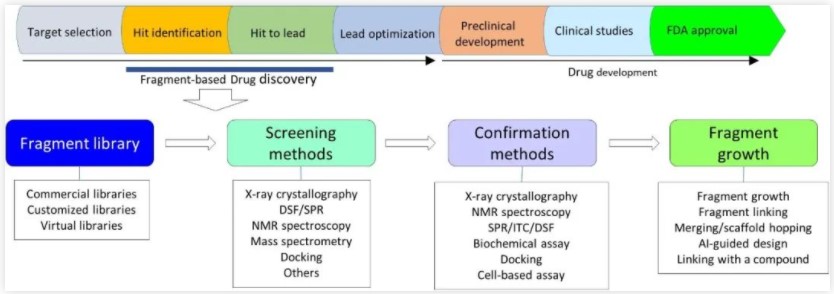
图9:片段的筛选、确认以及生长中使用的方法
— 总结 —
Copyright© 上海博璞诺科技发展有限公司 2016-2019 沪ICP备16015171号 中国化工网 全球化工网 生意宝 著作权声明
地址:上海市浦东新区张江高科技园区海科路100号9B楼四楼 电话:+86-21-20608178 传真:+86-21-20608171 联系我们

