详细内容
基于表型的药物发现在过去十年中重新兴起。
2022年5月30日,来自辉瑞全球研究、开发和医疗部门的Fabien Vincent等人在Nat Rev Drug Discov杂志发表文章,系统阐述和分析了表型药物发现近年来的发展情况、案例和对药物发现相关概念的影响,并讨论了表型药物发现面临的挑战。
.png)
主要内容编译如下。
摘要
在过去的十多年里,基于表型的药物发现方法在很大程度上被基于靶点的药物发现方法 (调控特定的感兴趣的分子靶点) 这种强大但简化的方法所取代。然而,将最初的理念与现代工具和策略相结合的现代表型药物发现在过去十年中重新出现。在此,我们讨论了表型药物发现方法最近取得的成功,并考虑了目前的挑战和解决这些挑战的方法。我们还探讨了这一领域的创新如何为下一代的成功项目提供动力。
前言
从历史上看,新药是通过观察其对疾病表型的治疗效果而发现的。随着20世纪80年代分子生物学革命的到来和2001年人类基因组测序的进行,重点转移到了特定的分子靶点。然而,自2011年以来,表型药物发现 (phenotypic drug discovery, PDD) 又重新兴起,因为人们惊讶地发现,在1999年至2008年期间,美国FDA批准的first-in-class药物中,大多数是在没有药物靶点假设的情况下凭经验发现的。
十年过去了,PDD作为一个领域正在逐渐成熟,成为学术界和制药业公认的发现模式,而不是一种短暂的热潮。这种持续的兴趣源于过去十年的显著成功,包括治疗囊性纤维化的ivacaftor和lumacaftor,治疗脊髓性肌肉萎缩症的risdiplam和branaplam,治疗精神分裂症的SEP-363856,治疗疟疾的KAF156和治疗特应性皮炎的crisaborole。
这并不是说PDD方法是解决制药行业生产力问题的灵丹妙药,表型筛选的利弊需要与验证靶点的分子方法仔细平衡。尽管PDD已经取得了成功,但许多历史上的案例都使用了高度复杂的疾病系统 (体内模型甚至是人类),而不是基于细胞的筛选,并且 (或者) 是偶然发现的结果 (图1)。药物重定向为这种状况提供了一个令人信服的案例:一方面有著名的基于偶然的临床观察的药物重定向的案例 (例如西地那非、米诺地尔、沙利度胺和金刚烷胺),另一方面则缺乏源于预先计划的临床化合物集筛选的获批的重定向药物。这就提出了一个关键问题:如何最好地利用表型筛选来发现新药。

图1:表型药物发现方法的案例
源自表型药物发现的药物案例,也说明了计划发现和意外发现的贡献。批准的药物是根据最初将化合物 (或药物本身) 与疾病联系起来的原始表型分析的类型列出的。所有来自细胞筛选的案例都是计划发现的努力的结果,而所有来自患者研究的案例都是基于意外的临床副作用,推动化合物的重新利用。
除了重新认识生理学和药理学的复杂性外,PDD还挑战了我们对什么是可药性 (具有不寻常的靶点和作用机制) 和什么是药物 (允许具有意外的化合物特性) 的假设。尽管在靶点识别、降低安全风险的步骤、以及为来自表型筛选的候选药物绘制临床路径方面仍然存在重要障碍,但功能基因组学、机器学习/人工智能和改进的疾病模型的应用正在带来令人兴奋的机会。
鉴于PDD和基于靶点的药物发现 (target-based drug discovery, TDD) 之间的主要区别、现在可用于PDD方案的新技术、该领域正在快速发展的情况,需要在工业界和学术界建立和分享最佳实践。尽管技术和文化方面的障碍依然存在,但PDD的重新利用已经开始改变我们对药物发现的概念化方式,并被证明是生命科学领域技术创新的重要试验场。在本文中,我们将重点介绍我们对PDD如何塑造药物发现相关概念的集体思考,并在最后讨论未来如何最大限度地发挥PDD作用的挑战。
PDD如何重塑药物发现相关概念
PDD在多个方面重塑了药物发现的相关概念,包括扩大可药用靶点的范围、重新审视多向药理学、重新审视"药物相似性"、重新认识靶点识别和项目进展之间的关系、改进靶点识别和MoA分析策略等。这部分还包括了对PDD产生化合物的临床开发问题的考虑。
扩大可药用靶点的范围
与TDD (基于分子靶点和疾病状态之间已建立的因果关系) 相比,PDD依赖于以分子靶点不可知的方式对疾病相关的生物系统进行化学询问。这种经验性的、生物学优先的策略提供了将治疗生物学与以前未知的信号通路、分子机制和药物靶点联系起来的工具分子,如以下四个示例所示。
第一个案例是丙型肝炎的治疗,这是一种由丙型肝炎病毒 (HCV) 引起的肝脏疾病。在过去的十年中,口服的直接作用的抗病毒药物的开发使HCV的治疗发生了革命性的变化,这些药物可以抑制HCV的复制,并清除90%以上的感染患者的病毒。HCV蛋白NS5A的调节剂,如daclatasvir,是这些直接作用的抗病毒药物的关键成分。NS5A对HCV复制至关重要,但没有已知的酶活性。NS5A的小分子调节剂,最初是使用HCV复制子表型筛选发现的。
另一个案例是囊性纤维化的治疗方法,囊性纤维化是一种进行性的、经常致命的遗传疾病,由囊性纤维化跨膜传导调节器 (CFTR) 基因的各种突变引起,这些突变降低了CFTR的功能,或中断了CFTR的细胞内折叠和质膜插入。研究人员使用表达野生型或疾病相关的CFTR变体的细胞系进行靶点诊断化合物筛选,确定了改善CFTR通道门控特性的化合物类别 (增效剂,如ivacaftor),以及具有意想不到的作用机制 (mechanisms of action, MoA) 的化合物:增强CFTR的折叠和质膜插入 (校正剂,如lumacaftor、tezacaftor和elexacaftor)。2019年,解决90%囊性纤维化患者的elexacaftor、tezacaftor和ivacaftor的药物组合获得批准。
第三,受沙利度胺有效治疗麻风病、调节多种抗炎细胞因子、抑制血管生成并在多发性骨髓瘤中显示活性等观察结果的启发,优化后的类似物来那度胺获得了FDA批准用于多种血癌适应症,并取得了巨大成功 (2020年的销售额超过120亿美元)。值得注意的是,来那度胺前所未有的分子靶点和MoA在获批后几年才被阐明。来那度胺与E3泛素连接酶Cereblon结合,重定向其底物的选择性,促进靶蛋白 (包括转录因子IKZF1和IKZF3) 的泛素化和随后的降解。此外,这种新的MoA现在正被密集地用于开发进一步的靶点蛋白降解剂,被称为"双功能分子胶"。
此外,在1型SMA (一种罕见的神经肌肉疾病) 中,由两个研究小组独立进行的表型筛选发现了能调节SMN2基因前mRNA剪接并增加全长SMN蛋白水平的小分子。这两种化合物都是接触SMN2第7外显子的两个位点并稳定U1小核糖核蛋白颗粒 (snRNP) 复合物,这是一个前所未有的药物靶点和MoA。其中一个这样的化合物risdiplam在2020年被美国FDA批准为第一个口服的SMA疾病修饰疗法。
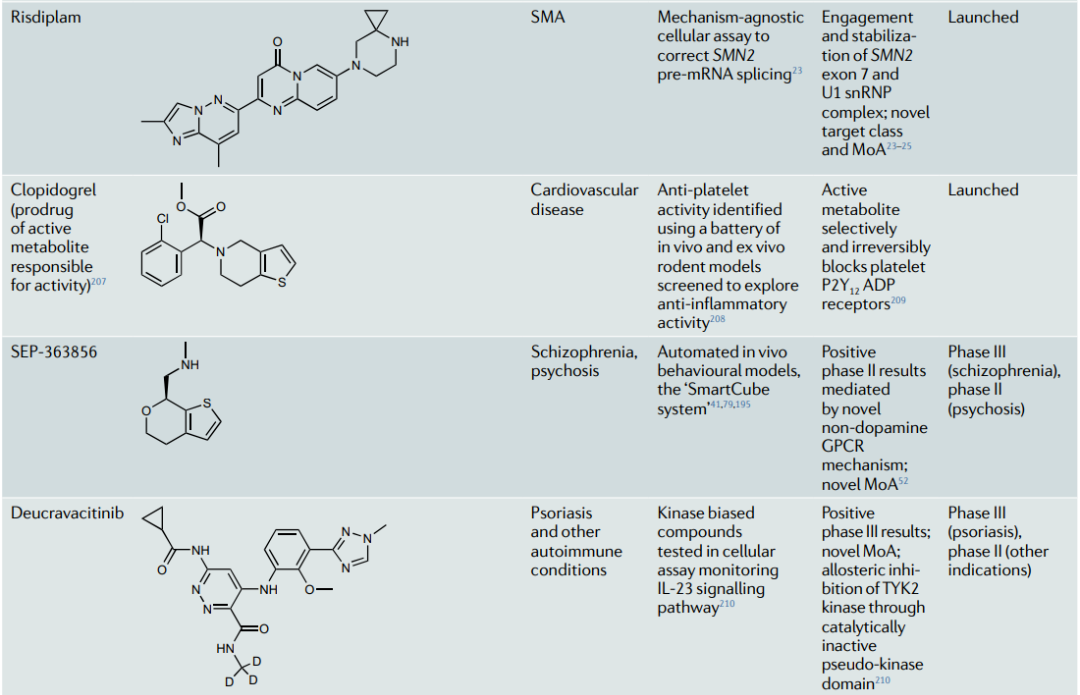
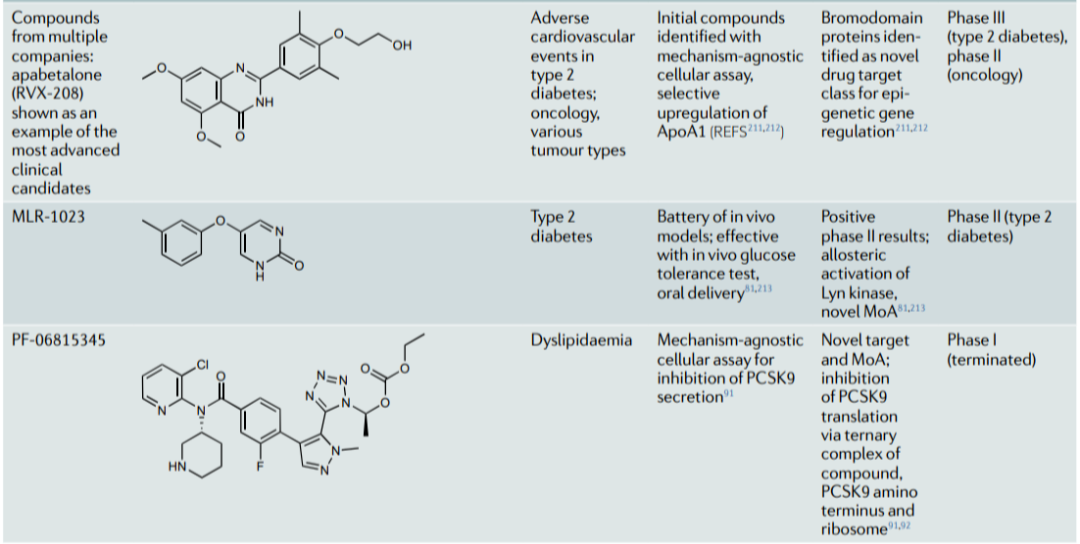
表1进一步介绍了最近通过表型筛选获得批准或处于临床阶段的化合物的案例。这些案例表明表型策略如何扩大了"可药用靶点空间",以包括意想不到的细胞过程 (前mRNA剪接,以及靶点蛋白的折叠、运输、翻译和降解) 和传统靶点类别的新MoA (伪激酶域抑制、异生激酶激活和掩盖的共价弹头),并揭示了新的药物靶点类别 (例如,溴模态域)。他们建议,当可以调节感兴趣的途径的靶点未知、或疾病表型和/或项目目标是获得具有差异化MoA的first-in-class药物时,应考虑表型策略。
表1:已获批药物和临床阶段化合物的表型来源

重新审视多向药理学
表型筛选提供了识别涉及多个靶点的分子的机会,即所谓的多向药理学 (polypharmacology)。在这种情况下,化合物的预期效果取决于靶点的组合。但是,其中可能包括不需要的靶点 (脱靶)。
在寻求选择性更强的药物的过程中,由于难以追踪非靶点所代表的所有生物功能,多向药理学传统上对化合物的优化不佳,容易产生潜在的副作用。然而,在治疗相关浓度下,大多数 (如果不是全部) 已获批准的药物都已知与多个靶点相互作用,这些靶点往往是副作用的基础,但也可能有助于临床疗效。
事实上,有人建议同时低药量调节几个靶点以达到"协同"的疗效,以减少副作用。一个典型的TDD案例是伊马替尼,这是FDA批准的第一个合理设计的激酶抑制剂,用于治疗慢性骨髓性白血病和其他癌症,目前正在临床开发中用于治疗近期发病的1型糖尿病。伊马替尼最初被认为是驱动慢性骨髓性白血病的BCR-ABL融合蛋白的抑制剂,它还表现出对c-KIT和PDGFR受体酪氨酸激酶等靶点的活性,这些靶点被认为有助于其在几种类型的癌症中的活性。
当只涉及单一靶点时,可能会产生抗药性。对于一些疾病 (如中枢神经系统和心脏疾病),基于单一靶点的方法的成功有限。一般来说,多靶点药物可能更适合于复杂的、具有多种潜在机制的多基因疾病,通常涉及与免疫或神经系统成分的相互作用。
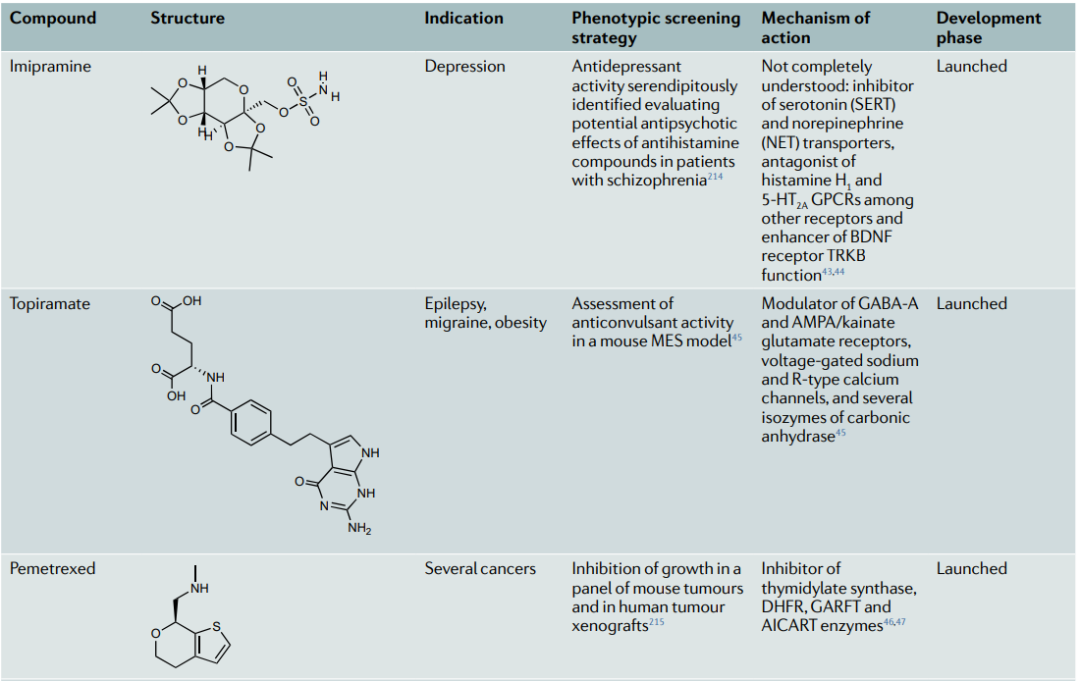
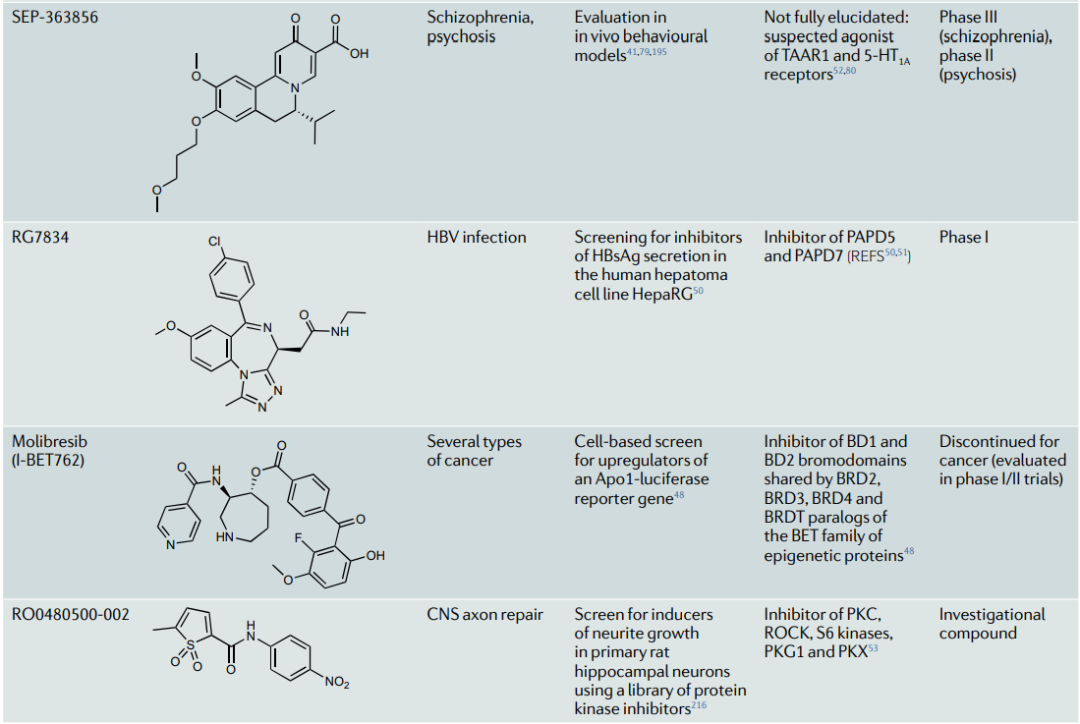
表型方法已经提供了许多药物和候选分子,这些药物和候选分子在MoA鉴定后显示出靶向多药理作用。代表性的案例包括咪唑,一种在20世纪50年代发现的三环类抗抑郁药,具有突出的多向药理学特征,可调节一些关键的中枢神经系统单胺转运器和受体。托吡酯,一种用于治疗癫痫和偏头痛的神经稳定剂,它能参与选择神经元受体、离子通道和酶;培美曲塞,一种被批准用于间皮瘤和非小细胞肺癌的叶酸抗代谢剂,它能抑制参与叶酸代谢和核苷酸合成的一系列酶。molibresib,一种第一代BET抑制剂,在I/II期试验中用于治疗几种癌症等。
不出所料,多向药理学经常发生在同一家族的蛋白质中,它们有共同的结构域或类似的底物或配体 (例如激酶、氨基G蛋白偶联受体和BET域蛋白)。
表2:多向药理学的代表性案例 (已上市药物、临床候选药物和研究性化合物中)
药物组合、工程化多靶点药物和多特异性抗体代表了简化的多向药理学方案,通常将两个部分结合起来,每个部分对每个单独的靶点具有选择性。将更复杂的多向药理学设计成一个实体,同时平衡候选药物所需的所有其他特性,这是一个复杂的过程,仍然是一项艰巨的挑战,尽管最近在分子对接、计算技术和人工智能方面取得了进展。
表型筛选提供了识别具有新的、无偏见的多向药理学特征的可能性。从药物发现的角度来看,表型筛选衍生的多向药理学可以利用日益强大的表型模型,结合先进的剖析、全局分析和计算技术,从偶然性发展到基于结构-活性关系的逆向工程方法,最大限度地减少潜在的安全风险,同时保持并最终优化表型活性,增加临床成功的机会。
重新审视"药物相似性"
现代表型筛选也可能有助于扩大新药所显示的分子特性的范围。具体来说,在过去的几十年里,随着TDD的出现,药物的分子量已经大大增加。历史上,较小的分子是通过表型方法发现的,通常使用动物模型,如布洛芬、米诺地尔和美满霉素 (图2)。这些分子完全在广泛接受的片段标准之内,如分子量<300 Da。
当代的案例包括利用癫痫模型发现lacosamide,利用中枢神经系统模型发现SEP-363856治疗精神分裂症,以及在多样化的体内模型中测试后将MLR-1023重新用于2型糖尿病和非酒精性脂肪性肝炎,并将富马酸二甲酯用于多发性硬化症 (图2)。
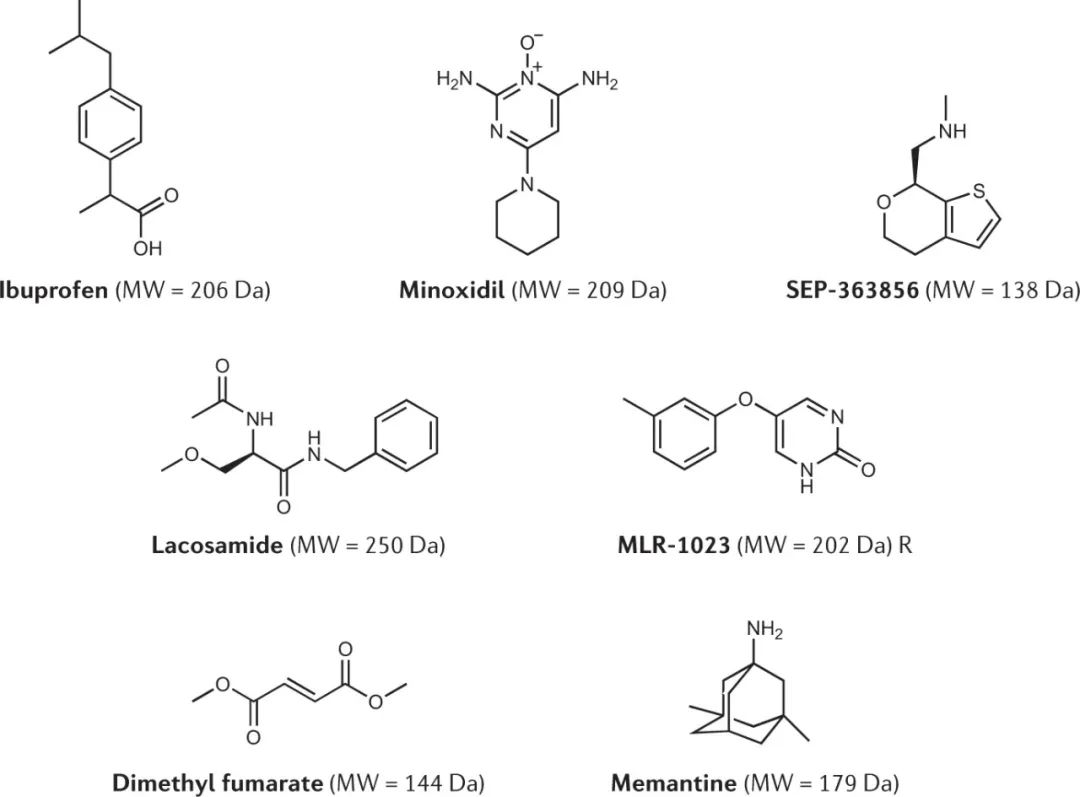
图2:来自表型筛选方法的低分子量临床候选药物和药物的化学结构
上述案例提出了一个关键问题:为什么现在被视为弱hit的、需要大量的优化的片段大小的分子,在体内疾病模型中却是活跃的,并在优化后能够获得类似大小的药物?
首先,已知片段大小的分子在针对特定靶点进行筛选时能提供更高的有效命中率,这是因为它们的尺寸小,适合结合位点的更多方向。如前所述,多向药理学是PDD药物的一个常见特征,也可能与此有关。而且,更小的分子在肠道通透性、通过细胞旁空间发挥作用方面已被证明具有优势,并且由于其较小的尺寸,它们对代谢酶的选择更少。尽管按照大分子的标准,许多这些片段大小的药物的剂量和相应的暴露量可能很高,但它们可能仍然显示出对其靶点的高配体效率,从而使它们能够安全地给药。换句话说,同样高的化合物浓度,对于分子量为200Da的化合物来说,与500Da的化合物相比,不会有同样的安全影响。
因此,存在一个机会,可以有目的地利用表型筛选的这种能力,来挖掘TDD方法中覆盖较少的化学和药理学空间。最近发现的SEP-363856、MLR-1023和lacosamide表明,使用体内动物模型进行表型筛选仍然可以取得成功。这种成功在很大程度上取决于模型的临床相关性。
展望未来,要考虑的第二个选择是调整这个概念,在复杂的多细胞类器官试验中筛选更少更小的分子。众所周知,尽管片段库的数量只有几千种,但却涵盖了一个很大的药理空间。测试片段库或分子量介于片段和高通量筛选中使用的典型化合物之间的分子,有可能允许覆盖一个重要的药理学空间,即使是低通量、复杂的三维检测。
一个相关的概念是使用共价片段 (含有可与蛋白质建立化学键的反应性分子的片段子集) 来识别新的靶点,并揭示调节靶点的化合物的化学起点。重要的是,共价片段得益于共价靶向 (持续的靶点接触) 和基于片段的筛选 (用一个小库广泛覆盖可配体的口袋) 的优势。例如,对初级人类T细胞中的共价片段的分析显示,T细胞激活的抑制剂通过不同的机制运作,包括直接的功能扰乱和/或蛋白质的降解。
关于靶点识别和项目进展之间关系的讨论
多年来,对于进入临床前的化合物,是否绝对有必要进行靶点鉴定一直是有争议的争论焦点。
靶点识别通常被视为导致一个简单的二元结果:要么靶点被识别,要么不被识别 (图3a)。在此,我们希望提供一个不同的框架来讨论这个话题。首先,重要的是要记住,靶点识别只是达到目的的一种手段,这个目的是为一个化合物系列在进入临床的道路上获得决策信息。因此,靶点识别只是实现这一目标的一种选择。此外,识别分子靶点并不等同于了解一个系列化合物的MoA,甚至可能在方案决策方面产生误导。
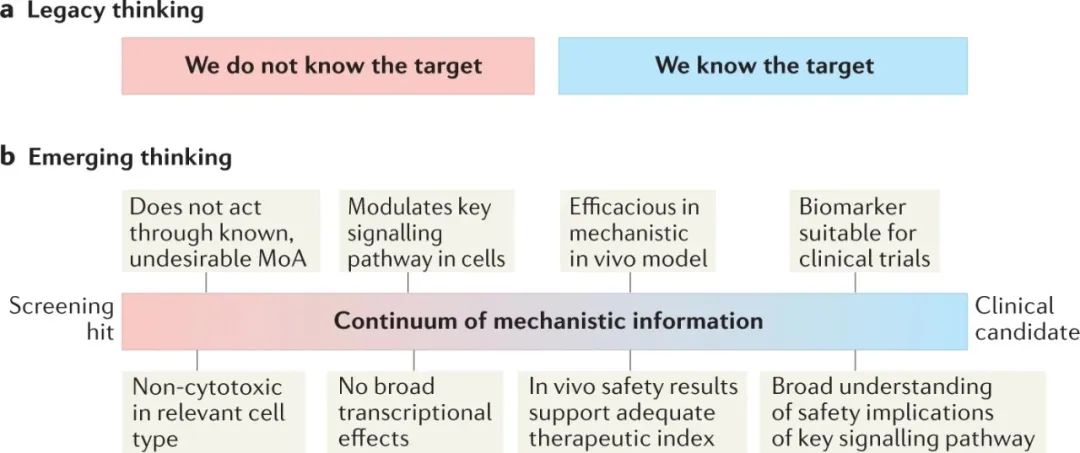
图3:表型药物发现中的靶点识别
a | 靶点识别有时被认为是源于表型筛选的候选药物取得进展的必要条件,也被认为具有简单的二元结果:靶点被识别或未被识别。b | 相反,我们建议可以获得一系列信息,这些信息可能涉及靶点识别的真正最终目标:帮助获得对安全性和转化的足够信心,以支持进展到临床。
例如,关于NS5A是HCV药物daclatasvir的靶点的文献,并没有合理解释其显著的亚纳摩尔细胞效力,也没有解释如何在化合物与蛋白质的比例低于1:1,000时获得疗效。同样,对于核糖体被确定为PCSK9分泌抑制剂的靶点,合理的反应应该是终止该计划,因为它提出了通过广泛的蛋白质合成抑制而产生重大安全问题的担忧。然而,包括蛋白质组学分析在内的进一步调查显示,由于核糖体、化合物和新生的PCSK9多肽之间形成三聚体复合物,其分子MoA对PCSK9 mRNA转录物的翻译提供了意外的特异性抑制。
作为靶点识别的替代方法,我们建议可以通过经验获得许多关于MoA的可行知识 (图3b)。实际上,细胞和体内试验可以提供无偏见的化合物评估,其读数与疗效和安全性有关,而现在经常采用"全能"方法的机理研究可能揭示与MoA有关的信息,如受化合物影响的特定生物途径。
在一个项目过程中,可以获得关于一个系列化合物的连续信息,而不是识别或不识别靶点的二元转换,这可能推动其在安全和转化方面积累的信心基础上进入临床。
靶点识别和MoA分析策略
亲和力 (或光亲和力) 富集结合化学蛋白质组学方法,或最近使用细胞热转移试验来确定与小分子化合物结合的蛋白质 (或蛋白质组) 是了解MoA的一种方法。但这可能还不够,甚至可能有误导性,如上面提到的PCSK9。
RNAi和CRISPR-Cas9的发展开启了筛选全基因组库的能力,允许以高特异性进行功能获得和功能丧失的研究。遗传扰动与化合物治疗相结合,提供了对MoA的进一步机理理解,并可能推动分子靶点本身的确定。一个大规模的努力是"癌症依赖性地图",其目的是利用PRISM方法在分子条形码细胞系中进行大规模平行化合物筛选,系统地确定遗传依赖性和小分子敏感性。
最近,提供化学扰动导致的生物变化的综合信息的分子分析方法在表型筛选的后续工作中发挥了突出作用。这种大规模分析工作的案例可以分为基因表达、细胞形态或生物标志物活性的测量。基于网络的综合细胞特征库 (LINCS)是美国NIH的一项共同基金计划,对细胞系在应对化学、遗传和疾病扰动时的变化进行编目。细胞画像使用形态学分析:从细胞的显微镜图像中提取定量数据,以确定样本之间的生物相关的相似性和差异。
这些大规模的分析方法,虽然产量较高,而且注重途径层面的信息,但在很大程度上依赖于参考数据库中具有类似表型的已知化合物。鉴于表型筛选背后的目的是发现新的MoAs,可能需要时间来建立知识库。然而,现在许多表型筛选是由特定的机理信息指导的。这种策略提供了一个关键的生物学框架,将基于上述工具的表型筛选命中的MoA的数据生成假设置于背景之中。
对PDD产生的化合物的临床开发考虑
在缺乏靶点信息的情况下,将PDD产生的临床前候选药物推向临床,对开发团队构成了若干挑战。简单地说,靶点识别提供了有价值的信息,既能消除安全问题的风险,又能预测和监测疗效。本节将介绍解决这些障碍的具体策略和案例。
“可转化链"驱动原始表型检测的机制、后续的临床前疾病模型和人类疾病的固有成分之间的分子水平关联,对于PDD项目在临床上取得成功至关重要。例如,HBV抗病毒剂RG7834在体外降低了含有耐受性病毒S抗原的非感染性膜质颗粒的分泌,因此捕获了人类疾病的一个基本 (和预后) 成分。治疗SMA的branaplam和risdiplam方案也为这一概念提供了一个强有力的案例。
在安全方面,靶点信息和伴随的有关其生理表达模式和作用的知识,经常被用来集中注意潜在的安全信号。这些可以在TDD项目的早期进行调查,以降低风险或迅速终止项目。
此外,研究人员可以获得更复杂的表型特征,并将化合物的特异性特征映射到斑马鱼或基于人类原生细胞的疾病系统中,用临床批准的药物产生的指纹进行验证。虽然这些系统不能完全再现在人类中可能观察到的安全问题的范围,但它们仍然提供了检测一些多器官安全问题的机会,同时允许测试大量的化合物。
在不了解分子靶点的情况下,来自PDD的候选药物可以而且已经过渡到了临床。例如,这些药物包括来那度胺 (2005年被批准用于多发性骨髓瘤,其MoA在2014年被阐明),lacosamide (2008年被批准用于癫痫,其可能的复杂MoA仍在调查中) 和RG7834 (最近进入I期试验,尽管靶点识别工作尚未成功)。
然而,在缺乏靶点信息的情况下,有必要确定能有效转化为人类患者的代用疾病生物标志物。
展望未来
表型筛选的代用表型
有时,表型筛选会带来一个难题。对于那些没有得到很好验证的治疗靶点的疾病来说,它显然是有价值的,而且对这些疾病的理解和特征也可能相应地不那么充分。然而,疾病知识对于设计具有相关体外或体内生物系统、刺激和读出的表型测定以及建立所需的转化链至关重要。就像它们被用来定义小分子的MoA一样,基因表达和细胞形态等高维信息可被用来定义疾病的代用表型,作为表型检测的读出值。
基因表达分析已被用于定义疾病状态,如癌症中基因组改变引起的疾病。例如,高通量mRNA谱被用来根据其功能影响对肺腺癌中发现的等位基因进行分组。
细胞形态表征,包括细胞区室的形状、大小、强度和质地,已被证明会因扰动 (无论是小分子还是疾病相关的等位基因) 而改变。目前,LINCS门户网站已经纳入了来自Cell Painting和Drug Repurposing Hub的此类数据,报告了1,571个化合物的Cell Painting数据 (其中92%映射到人类蛋白靶点或分配了MoA标签)。这些数据可用于定义由已知MoA的药物逆转的表型。
与基因集富集分析类似,Nassiri等人开发了一个细胞形态富集分析,以评估转录组改变和细胞形态变化之间的关联,强调转录和细胞形态之间的相互依存关系可以与疾病状态联系起来。
本节中提到的高维分析数据集的储存库已经存在于公共领域。对于药物发现来说,如何最好地利用这些数据来定义可以逆转的替代疾病表型作为治疗效果的指标,是该领域的下一个重大障碍之一。一些制药公司已经开始使用这种方法作为一个筛选平台。
从2013年开始,Recursion Pharmaceuticals可能是第一个通过Cell Painting,使用疾病的代用特征,来筛选替代性疾病逆转特征的工业化努力。 他们的主要重点是对罕见的单基因疾病进行药物重定向。
人工智能和PDD
人工智能的应用在药物发现和开发的多个领域已被广泛接受,包括药物设计、蛋白质折叠、化学、毒性预测和药物重定向。这种方法的指数级应用速度依赖于机器学习算法识别模式的能力,并从它们与某些参数 (如效力和选择性) 的关联中学习。
使用关键词”深度学习"、"人工智能”和”表型”或”药物发现”在PubMed上查询已发表的文献,会发现两种不同类型的论文:一种是将机器学习分类器应用于大量化合物或化学结构的集合,另一种是将分类器应用于表型检测衍生的特征。绝大多数发表的文献属于第一类,使用基于图或基于深度学习的算法,应用于大量的化合物或化学结构的集合,以及先前筛选中产生的相关药理学数据。
相反,将机器学习分类器用于表型衍生特征的努力仍然很少,尽管这显然是一个诱人的应用,可以揭示隐藏在对普通观察者来说显然是混乱的模式。根据定义,PDD依靠表型模式的变化来识别和优化分子,而对生物靶点或MoA知之甚少或一无所知。在细胞水平上对药物诱导的扰动进行表征 (例如,使用细胞画像),表明亚细胞特征指标可用于对化合物和基因扰动进行聚类和分类。机器学习在特征空间定义不明确的情况下特别有用,因此对PDD有明显的帮助。
机器学习与PDD的整合也有其他机会。例如,表型筛选和机器学习可以结合起来,从杂乱的化合物集合中提取靶点信息。
有两项研究强调了将机器学习应用于PDD的变革性潜力。虽然其他人已经将深度学习应用于抗生素的发现,但Zoffmann等人使用了一种结合了高通量成像和基因组学的方法,结合机器学习驱动的数据集分析,有效地缩小、比较和预测化合物MoAs。在这项研究中,机器学习的应用可以在特征空间中定义化合物的”原型",使化学家在进行化合物优化工作的同时,不断监测这些修改如何影响类似物的MoA--这是与传统PDD的一个重大区别。杨森公司的研究人员将这一概念提升到新的水平,将高通量成像与机器学习结合起来,建立了一个概念证明,即来自特定细胞检测的图像可以支持对一系列生物检测的活性预测。
生理和疾病相关检测系统的进一步发展
一个PDD项目是成功还是失败,取决于”可转化链" (如上所述) 的内在强度,该链将一开始的主要表型检测与最后的病人疗效联系起来,中间往往有一个动物疾病模型。Scannell和Bosley对生物制药行业的分析表明,研发效率的下降可能是由最具预测性的疾病模型逐渐耗尽造成的,而新的疾病相关模型的创建速度可能是研发生产力的主要制约因素。综上所述,继续开发现实的疾病相关检测方法,并验证其临床转化能力,对未来的PDD工作具有至关重要的意义。
幸运的是,不同学科的进展,如干细胞生物学、功能基因组学、生物工程/微细加工和仪器/数据分析,已经融合在一起,为开发潜在的疾病相关检测系统提供了广泛的实验平台。技术进步包括但不限于模型生物的平台方法、高通量体内哺乳动物药理学、使用高保真Cas9方法来调节基因调控/结构、使用三维基质/微流控系统和器官芯片等。
与设计疾病相关的生物模型有关的实验变量的复杂性和数量都很重要。不幸的是,在一个模型中重现相关病人生物学的所有方面,充其量是一个理想的目标。更现实的是,研究通常集中在再现特定的疾病特征,这些特征被认为是模型价值的关键。即便如此,这些通常是复杂的系统,因此需要大量的开发、优化和验证工作。与分子靶点验证一样,只有在发现和临床数据之间建立一致的情况下,疾病模型才应被认为是可以转化的。
在大数据时代,整合来自大型人群的真实世界患者记录 (例如英国生物库和FinnGen) 及其组学数据,也可用于帮助建立和验证模型系统。利用监测α-突触蛋白基因表达的体外表型筛选作为帕金森病模型,Mittal等人在药物重定向筛选中确定了β2-肾上腺素能受体调节剂。重要的是,体外模型随后通过对400万份病人记录的分析得到了验证,该模型将使用β2-肾上腺素能受体激动剂或拮抗剂与帕金森病发病风险的降低或增加分别联系起来。
结束语
PDD通过识别药物、靶点和MoA证明了其潜力,在许多情况下,使用基于靶点的方法是不可能发现这些药物、靶点和MoA的。当缺乏关于疾病病理生理学的分子信息时,这种策略提供了一条通往新型疗法的道路,提供了进入由蛋白质组和任何其他生物大分子和疾病基础细胞过程所代表的未开发的”黑暗生物物质”的机会。
表型模型的临床转化能力对于PDD的成功至关重要。
这里强调的关键方面包括:发现只有PDD才能获得的新MoA、需要能更好地再现复杂疾病的病理生理学的表型模型、多向药理学提供的机会以及使用比传统分子小的化合物库的优势。应用于疗效/安全性评估的生物活性分析和MoA表征方法的增加,以及日益强大的计算技术,已成为充分挖掘PDD潜力的关键。
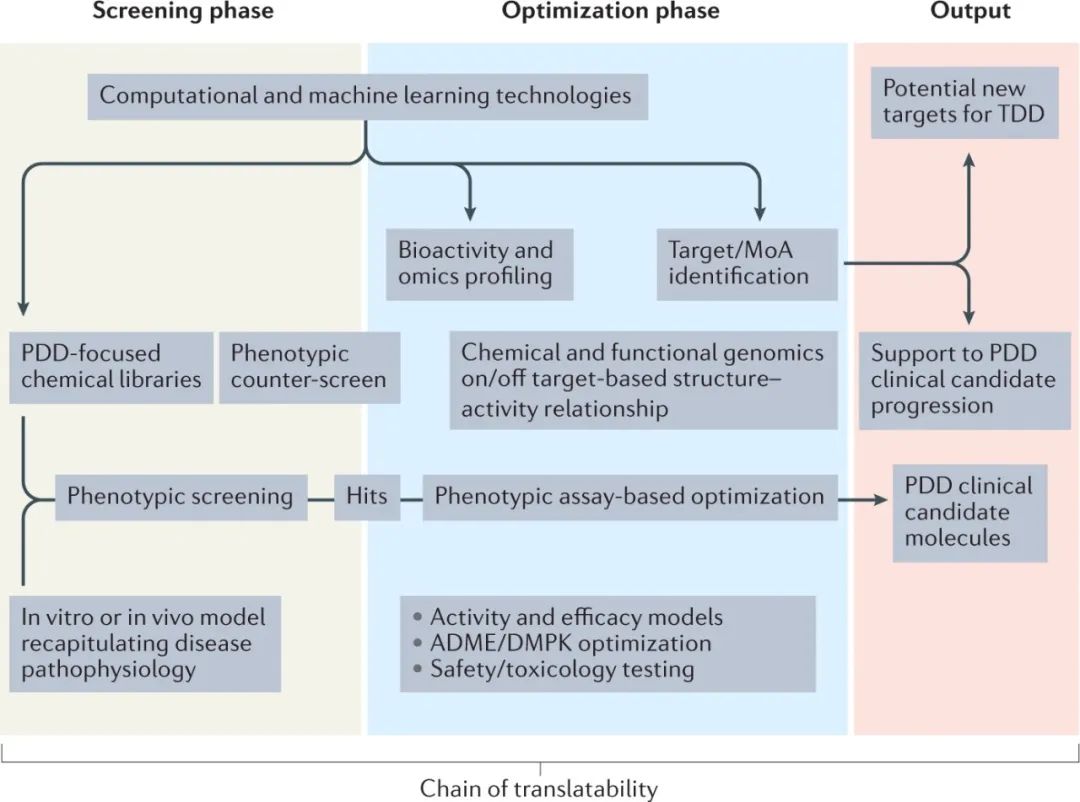
图5:工业化表型药物发现过程的示意性概述
目前的挑战是如何合理地结合这些关键方面,使PDD”产业化"。鉴于该领域的指数级增长,我们相信,机器学习 (尤其是深度学习) 的增加应用将有助于PDD的有效实施 (图5)。我们的愿景是,PDD的产业化,加上经过十多年的”现代”PDD实践所积累的丰富经验,将有助于实现更有成效的药物发现过程,将基于靶点和基于表型的方法无缝整合。
参考资料
Vincent, F., Nueda, A., Lee, J. et al. Phenotypic drug discovery: recent successes, lessons learned and new directions. Nat Rev Drug Discov (2022). https://doi.org/10.1038/s41573-022-00472-w
Copyright© 上海博璞诺科技发展有限公司 2016-2019 沪ICP备16015171号 中国化工网 全球化工网 生意宝 著作权声明
地址:上海市浦东新区张江高科技园区海科路100号9B楼四楼 电话:+86-21-20608178 传真:+86-21-20608171 联系我们

