详细内容
转自:饶议科学 2023-08-09 07:31 发表于广东
【试想,如果吃药就可以一时消除一切或大部分痛疼,这个世界会多么神奇。2023年8月3日,《新英格兰医学杂志》的论文,不仅让我们浮想联翩,而更应该饮水思源。
以前镇痛非常有效但是作用于中枢的吗啡,有很强的成瘾性。新药根本完全不同于旧的中枢镇痛药。也不同于传统的外周镇痛药如阿斯匹林。阿司匹林是抑制外周部分痛觉信息等产生。新药依据人类遗传学和分子神经生物学,抑制外周痛觉传导通路。
如果出现非常有效的、无成瘾性的外周镇痛药,很可能不是某些病人需要它,而是全世界每一个人平时都愿备用。新药在药效和副作用都还没有达到人们的希望,但提供了进一步改进的基础,为新型镇痛带来了曙光。
甚至,不能断定它不可能触动我国生物医药产业界放弃跟风“媒体常见病”,而让一些没学过医的从业人员认识到人民对医药的需求面之广远超过一般媒体报道】
中文媒体,经常哗众取宠,把通常的研究论文,都当成突破来进行报道,久而久之,恐怕读者耳朵都要起老茧了。
如果按媒体那样报道生物医学方面的突破,全世界的疾病早就没有了。
科学确实有突破,只是不多,而且不是由媒体能够定义,只有同行科学家能够估计,而且科学家也不能百分之百准确。我自己的准确率不算低,但也不到百分之百,也可以出错,发表意见的时间越快越容易出错。所以,本文也有百分之五的出错率,请事先知悉,免得事后后悔。
痛觉是保护人类的必不可少的感觉。如果没有痛觉,人就难以健康成长。事实上,有非常少的人,因为先天基因突变,缺乏痛觉。一般来说,他们伤痕累累,而且会误解自己的特殊性。有记录,缺乏痛觉的孩子从高楼跳下去,因为把不痛的奇迹误认为预计不死。但,虽然不痛,照样会死:迄今有极少数不痛的人,但没有一个不死的人。
所以,痛觉不仅仅是一般人抱怨的疾病症状,它也有帮助人类的一面。
对痛觉的理解,有一个重要的突破是发现有专门参与痛觉传导的钠离子通道蛋白。这一突破十几年之后,终于有以钠通道为靶点的新药在人类显示一定镇痛效果,特别是有3万倍的靶点选择性。这为出现疗效更好、副作用更低的外周镇痛药带来了崭新的希望。
有正常的神经才是人
人体最重要的是神经系统。其他动物的神经是否最重要,有点说不清楚,例如相对于心血管系统。但对人来说,最重要的肯定是神经系统:假如所有系统都工作,但只要神经系统不工作,我们就不是人。
我们每一个人最关键的不同也在于其特有的神经系统。
神经系统最重要的细胞是神经细胞(还有其他例如胶质细胞),也称为神经元。神经元接受和产生神经冲动,通过神经纤维传导。
神经冲动的离子基础

1781年,意大利科学家Luigi Galvani发现神经纤维可以产生电。
1838年,意大利科学家Carlo Matteucci检测到神经纤维(和肌肉)上的电流。
1841年,德国科学家Emil du Bois-Reymond记录到神经纤维上的负变化,当时称为动作电流,以后着重“动作电位”。

1850年,德国科学家Herman von Helmholtz首先记录到动作电位的传导速度,否定了神经冲动速度神速不可测量的观点。神经系统无论多么神奇,仍然是遵循物理规律的客观事物。

1902年,德国犹太生理学家Julius Bernstein提出神经纤维静息状态时,细胞内钾离子外流,维持细胞内负外正的极化状态。但他误认为动作电位期间,细胞膜对所有阳离子的通透性都增加,导致细胞膜电位为0。

1902年,德国生理学家Ernest Overton提出细胞外的钠离子对于神经的兴奋很重要,也就是钠离子内流可能是动作电位的基础。
1936年,英国牛津大学的John Young使用枪乌贼的巨大神经纤维进行研究。美国哥伦比亚大学的物理学家Kenneth Cole访问Young后学到用枪乌贼巨神经。
1939年,Cole和学生Howard Curtis研究巨神经发现动作电位期间,膜电阻减小,膜电导增加。

英国剑桥大学生理学家Alan Hodgkin访问美国纽约的洛克菲勒大学,得以结识美国科学家Peyton Rous的女儿,以后结婚。Hodgkin还到纽约的哥伦比亚大学与Cole交流,之后跟着Cole在海洋实验室研究枪乌贼的巨神经。
1939年,Hodgkin与学生Andrew Huxley发现动作电位发生时,膜电位并非0,而是逆转为膜外负、膜内正。因为与静息时的内负外正相反,所有称之为去极化。
1939年,Cole和Curtis有同样发现,但发表在1940年。
1942年,Curtis和Cole证明静息电位由钾离子外流造成。
1946年至1947年,Cole实验室发明的后来被称之为“电压钳制”的技术,由其实验室的Marmont单独发表。八十多年来,一般人(包括绝大多数教科书)不知道电压钳制技术的起源,错误地归功于Hodgkin和Huxley,而且找不到应该引用的原始文献(Marmont,1949)。
1947年,Hodgkin和Huxley实验证明,动作电位之后恢复膜电位的复极化过程是由于钾离子外流。
1949年,Hodgkin与犹太学生Bernard Katz证明动作电位是由于细胞外的钠离子流向细胞内所导致。
此后,一般来说,所有的神经纤维的冲动发生和传导都需要钠离子内流。如果没有钠离子内流,就没有动作电位,神经系统就不能传导信号。
细胞膜是脂质双层膜。而细胞膜上面有特别的离子通道,这些离子通道是蛋白质。通透钾离子的是钾通道蛋白,通透钠离子的是钠离子通道蛋白。
1984年,日本科学家沼正作发现第一个编码钠离子通道蛋白的基因。

1987年,美籍华裔科学家叶公杼和詹裕农首先通过研究果蝇的突变基因,发现编码钾通道蛋白的基因。

1998年,美国科学家Roderick McKinnon解析第一个离子通道蛋白质(钾通道)的结构。

区分中枢与外周镇痛的关键实验
痛觉对于保护人类非常重要,没有痛觉的人难以长期健康生活。先天缺乏痛觉的人会出现认知偏差,难以活到成年(Dearborn, 1932;Landrieu, Said and Allaire,1990;Nagasako, Oaklander and Dworkin,2003;Cox et al., 2006)。痛觉又经常让人们难受,人们经常希望能够有镇痛的方法。
一般来说,伤害性刺激由于其物理、化学性质可以特异地激活痛觉神经末梢,由痛觉神经传导信息至脊髓,上传至脑,经过引起痛觉(Basbaum et al., 2009)。通过长期的研究证明,痛觉传导通路与其他体躯感觉如温度、触压、痒的传导通路是分开的,而不是其他感觉过度强烈后成为痛觉(Perl, 2007)。对于痛觉传导神经非常重要的钠离子通道蛋白Nav1.7及其基因SCN9A已被发现,其功能增加的突变导致自发或轻微刺激就出现痛觉(Yang et al., 2004),而其缺失导致无痛(Cox et al., 2006)。因为它是痛觉神经特异的,所以其突变不影响稳定、触压等其他感觉。
镇痛的药物可以在外周发挥镇痛作用、也可能在中枢发挥镇痛作用。

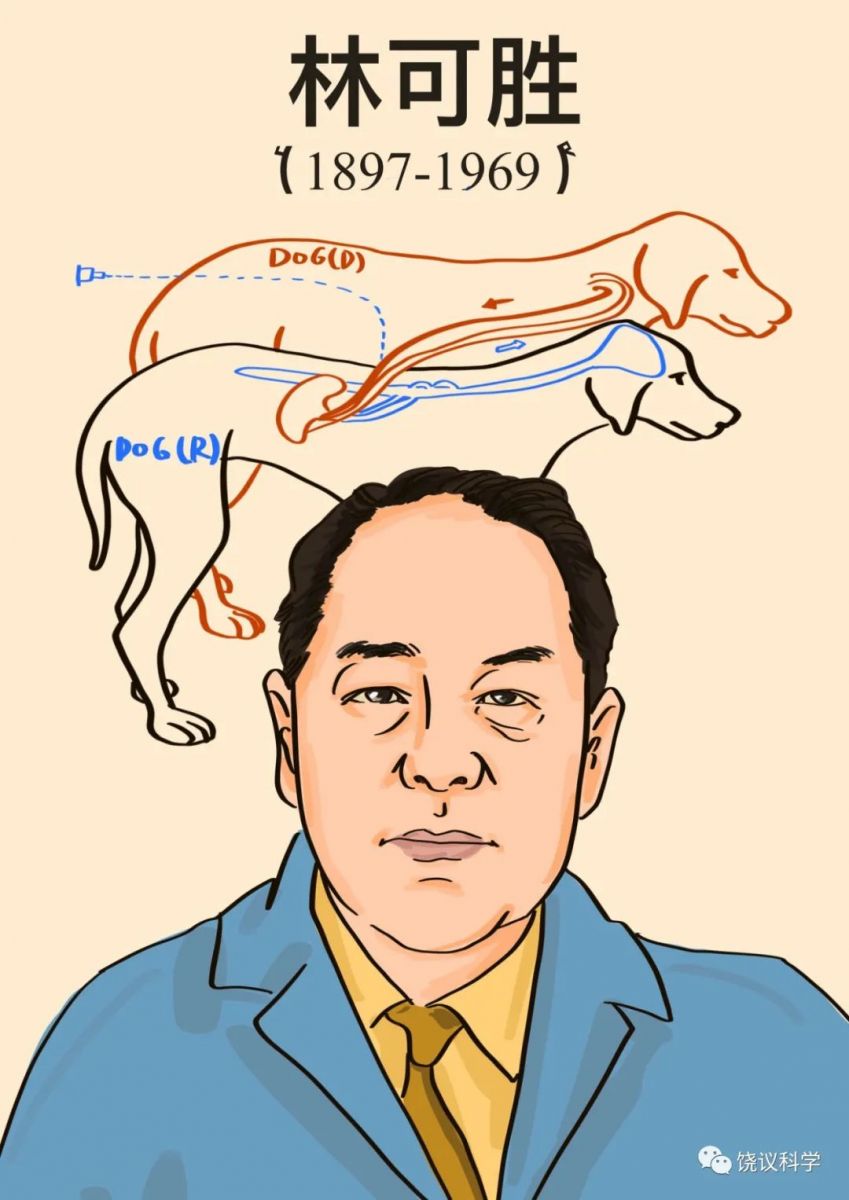
1964年,当时在美国Miles药厂工作的林可胜(Robert KS Lim, 1897-1969)发明了区分药物是在中枢还是在外周起镇痛作用的方法(Lim et al., 1964)(详细见明天文章,镇痛药简史)。
林可胜通过这一优美实验设计建立了鉴定中枢和外周镇痛的一个标准模型,可以鉴定很多药物的外周、中枢差别(Lim,1968)。同时,它证明阿司匹林镇痛作用的位点在外周,是阿司匹林研究的一个里程碑。
阿司匹林是如何在外周发挥镇痛作用的呢?其生物化学机理是抑制前列腺素的合成(Vane, 1971; Smith and Willis , 1971),而有些种类的前列腺素可以刺激痛觉系统的外周神经末梢,因此阿司匹林能够缓解一部分疾病的痛疼。
林可胜的研究结果也支持了此前中国国内科学家邹冈和张昌绍的发现:吗啡在中枢神经系统发挥镇痛作用。

标记的神经纤维通路
对于各种外周感觉来说,由不同的刺激,兴奋不同的神经末梢,再通过神经纤维传导到脑的不同区域。脑对外周信号的加工后,产生各种感觉。
与所有感觉如视觉、听觉、触压、温度一样,痛觉也需要神经纤维传导。
传导不同感觉的神经纤维,除了末梢能够感受不同的刺激外,是否纤维还有不同。一系列研究证明,不同的感觉,有不同的神经纤维传导。其中比较特别的是,传导冷感受和热感受的纤维居然也不一样。
痛觉特异的钠离子通道蛋白
不同离子的通道蛋白不一样,而同一种离子有多种通道蛋白。
没有先验的理由,可以预计传导不同感觉的神经纤维需要不同的离子通道。
所以,发现有特别的钠离子通道蛋白只参与传递痛觉,令人惊讶。
人类遗传学的贡献
人类遗传学是指人进行遗传分析、遗传学研究。
对于一般人来说,研究人类疾病与基因的关系,就是常见的人类遗传学。
1970年代末开始,世界上逐渐发现越来越多与疾病相关的基因。而我国被一群水平很差的人误导,在一群不懂的行政领导支持下,我国对基因测序投资巨大,而真正重要的发现人类疾病基因很少。
中国的人类基因研究为什么弱?
核心技术追问:中国的人类基因研究为什么弱?
在中国罕见参与发现人类重要疾病相关基因的大背景下,也有几个很好的研究工作。其中之一是在痛觉相关基因的发现。
中国发现人类疾病基因的两个重要贡献
引用我在2018年的微信公众号所写(在2018年之前也写过几次,不在微信上):
“痛觉是非常重要的感觉,没有痛觉的人,难以存活很久。但世界上,真有缺乏痛觉的人,当然极为罕见。科学上报道第一例缺乏痛觉的人是1932年,迄今也不过十几例。在巴基斯坦发现三个遗传痛觉缺失的家系,都是近亲结婚的家庭。其中一位小孩14岁不到就去世,原因是他没有痛觉,平时经常在街头表演刀刺自己、火上行走,他不能感到痛,但这并不意味着他不受伤害,而是受了伤害不感到痛。一个人如果从来不感到痛,他对世界的理解就会出现偏差。这个孩子后来跳楼,当然他确实不痛,但与常人一样,跳楼会死,他因此去世了。
还有一种病,称为“红斑肢痛症”,患者会自发或者受到轻微的刺激就出现肢体剧烈疼痛、皮肤红、灼热。Waxman的书名,可以视为是“灼热痛”和“火上走”双关语。1966年,美国Mayo医院的医生发现阿拉巴马州有一个较大的家系,5代51人中19位有自发疼痛。1992年,阿拉巴马大学医学院的教授发现同一家系更多病人,29人患病。荷兰遗传学家联系美国阿拉巴马州的医生,获得这一家系多个病人和正常人的DNA样本,进行分子遗传分析,发现病人是因为2号染色体特定段落出现突变,这段含约50个基因,但他们不知道是哪个基因导致自发疼痛。
北京大学第一医院皮肤科的杨勇,发现一家三代有“红斑肢痛症””。他进一步分析了这50个基因,发现包含有一组钠通道基因,而其中SCN9A比较特异的表达在外周神经,与伤害感受有关,是最可能的致病基因。后面的工作证实了他的猜想,他们发现SCN9A基因在家系中每一个病人都有相同的突变,而正常人没有。他们进一步分析散发病人,有一位“红斑肢痛症”病人也在同一基因有另外一个突变。杨勇因此提出SCN9A突变是“红斑肢痛症”的致病原因。这一结论于2004年发表在《医学遗传学杂志》。后续杨勇与Waxman合作,阐明了疾病的发病机制:突变导致SCN9A功能增强,从而对伤害刺激更敏感。

2006年,英国遗传学家在杨勇研究的基础上,专门研究巴基斯坦三个先天缺乏疼痛的家系,三家有六个“病人”,完全没有痛觉。英国遗传学家发现他们都是因为发生SCN9A的功能缺失型突变而失去痛觉感知能力。这些缺乏痛觉的人身体很多伤害,包括小时候自己咬坏了自己。他们都有正常的触觉、压觉、温度感觉、本体感觉。文章发表在《自然》,当年我是看到这篇文章,再追踪得知北大第一医院杨勇老师做出的关键一步。当然,2004年杨勇的《医学遗传学杂志》论文比英国遗传学家的《自然》论文更重要,但《自然》杂志的读者多。
影响痛觉的基因不止一个,但确定SCN9A与痛觉的关系非常有助于理解痛觉传导。
SCN9A基因编码Nav1.7蛋白质,这是钠通道蛋白质的一个组分(专业称为“亚基”)。杨勇的发现,首先表明Nav1.7钠通道是痛觉特异的离子通道。杨勇研究的病人实际是基因突变后,Nav1.7钠通道功能增强,导致自发疼痛。而英国遗传学家研究的巴基斯坦疼痛缺乏的病人所含的遗传突变,导致Nav1.7钠通道功能缺失。所以,两项工作相辅相成,证明Nav1.7钠通道对于疼痛传导非常重要。但杨勇是第一个发现Nav1.7钠通道对疼痛的重要性。杨勇的工作既有国外工作的基础,也有国外工作的继续,但杨勇的工作是关键一步。这一工作既揭示了一个疾病的罹患基因,同时提出了疼痛的一个特异机理,而且提示可以寻找抑制Nav1.7钠通道的药物而治疗疼痛。所以,杨勇的工作意义很大”。
最新研究结果
确定Nav1.7钠离子通道蛋白的痛觉特异性后,全世界药厂扑上去寻找可以抑制它的分子。
后来发现Nav1.7也参与交感神经系统,而Nav1.8和Nav1.9两个钠离子通道蛋白等也特异参与外周感觉传入。所以,这Nav1.8和Nav1.9钠通道蛋白成为全世界大小药厂的热点。
2023年8月3日,美国的《新英格兰医学杂志》发表文章,Vertex药厂发明的VX-548,对Nav1.8有高度选择性(3万倍),在多中心临床试验后显示对人的痛疼有抑制作用,但还有副作用。
这一结果,显示痛觉分子机理的基础研究已经在临床上初现曙光。
这一曙光,是物理学家、生理学家、遗传学家、药理学家、临床医生前赴后继上百年研究的结果。
如果这条路能够走下去,那么,不仅可以探讨特异性的化学药物,可能还有其他药物,包括基因编辑的可能性。

同期《新英格兰医学杂志》发表了美国耶鲁大学Waxman教授对这一结果的述评,也值得阅读。不过,他对于钠通道是谁提出、谁发现的历史搞错了。这是一个很常见的错误,一般人都忽视其他科学家而说是Hodgkin和Huxley,因为他们获得了诺奖、而且1950年代有几篇在神经生理学内部脍炙人口的、每个美国神经生物学研究生都必读的论文。

结构生物学对药物设计的指导性意义
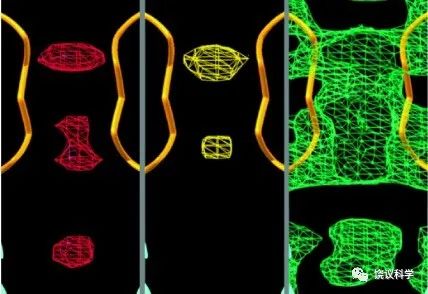
在遗传学确定钠通道对痛觉的重要性之后,结构生物学可以为设计药物提供指导性的信息。颜宁实验室是世界上解析钠通道蛋白的权威。Nav1.8的蛋白质三维结构也是她们于2022年解析的。

其他一堆钠通道的结构(包括1.7)都是她们解析的,估计她们最有设计相关药物的洞察力。
当然,中国完全真正全新的药物设计目前还比较少,以仿制为主。但今后可能会在真正创新方面逐渐赶上,特别是有科学基础的时候。
微信讨论科学
微信中得知,有机化学家参与了VX548的设计。
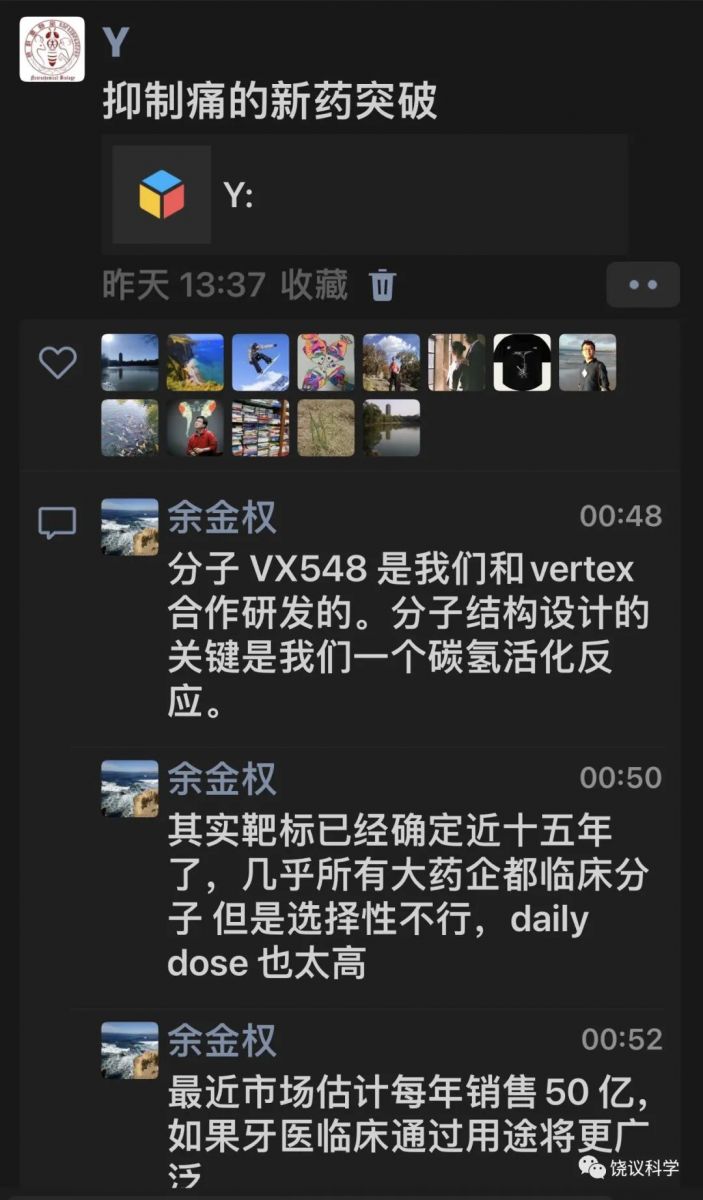
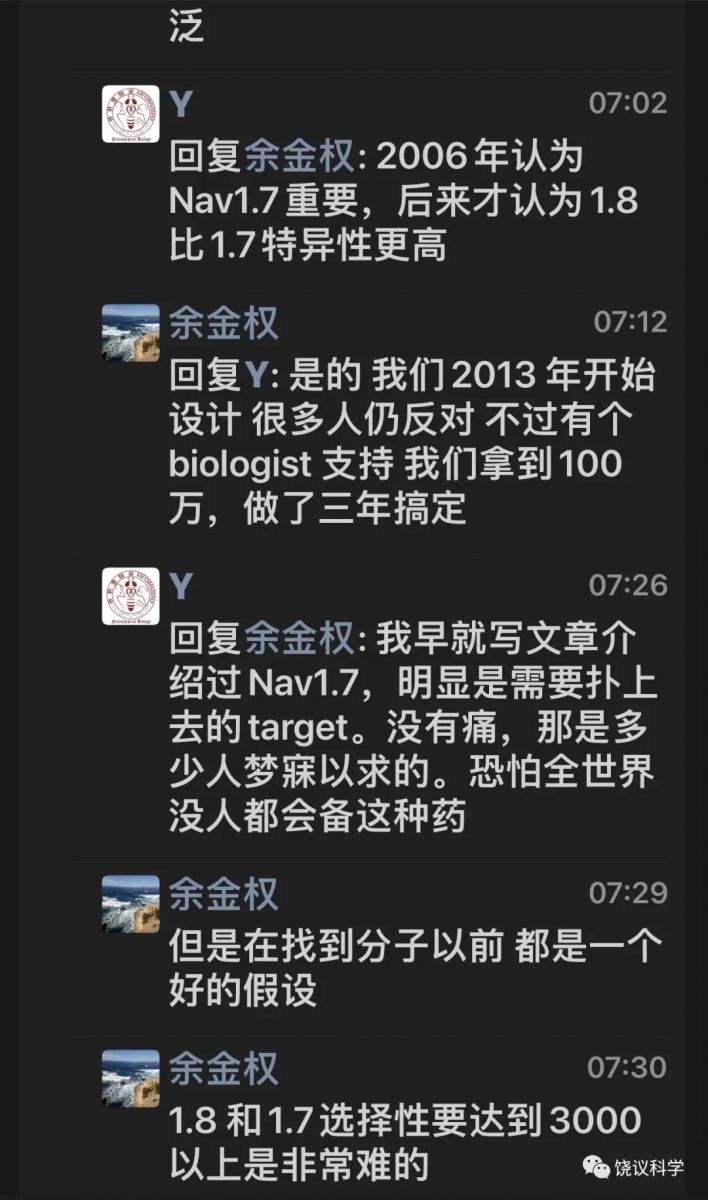
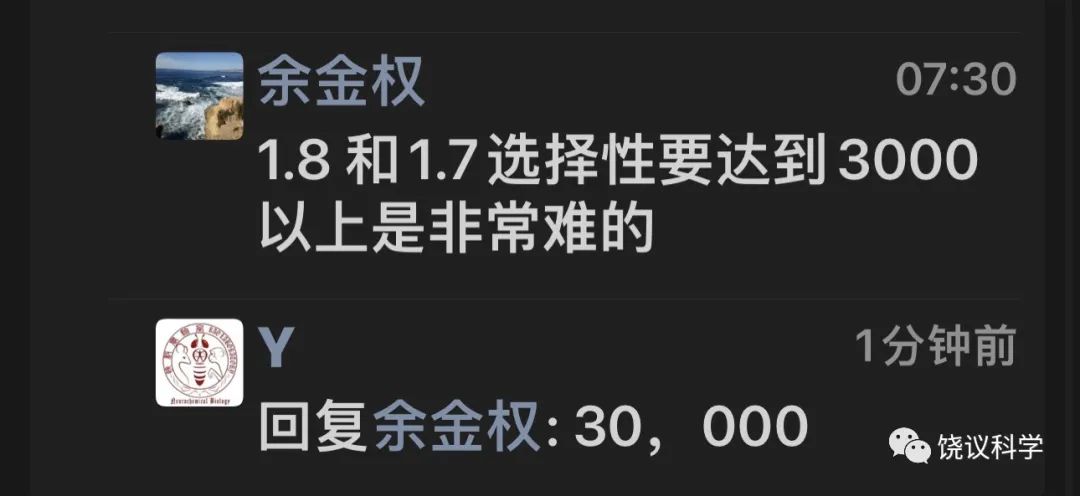
著名化学家余金权介绍:
“研制取代有毒瘾的镇剧痛的小分子药堪称一部血泪史,多少药企豪门在过去二十年先后倒在一二期临床上。我们2013 年啟动和Vertex的合作,用碳氢活化编辑有针对性的创造新的四氢呋喃化学空间,解决了钠离子通道1.8和1.7,1.9,1.4,1.5 的选择性难题。VX548 对1.8的选择性高达30,000,让其他临床分子望尘莫及。上周临床医学权威杂志新英格兰医学总结了VX548 的三期临床结果。
早期合作的论文不受/待见:1.8钠离子通道很难成药;导向碳氢活化没有非导向高大上。最后被多个杂志拒稿后发表在欧洲化学,2016,22,4748。当然回头看,该论文价值连城。我斗胆说一句,我们化学界马户何其多,马骥何其少!
我也特别希望该分子用于牙医的三期临床也获得成功,让那些喜欢嗐喷的马户们在拔牙时体会到导向碳氢键的意义和用途”。
颜宁也许有更多想法,也许余金权与颜宁合作会设计、合成出更好的外周无成瘾性镇痛药?
部分参考资料:
Tsou K and Jang CS(邹冈,张昌绍)(1962) 脑室内或脑组织内微量注射吗啡的镇痛效应.生理学报 25:119-128.
Tsou K and Jang CS (1964) Studies on the site of analgesic action of morphine by intracerebral microinjection. Sciencia Sinica 8:1099-1109.
Lim RK, Guzman F, Rodgers DW, Goto K, Braun C, Dickerson GD and Engle RJ (1964) Site of action of narcotic and non-narcotic analgesics determined by blocking bradykinin-evoked visceral pain. Archives Internationales de Pharmacodynamie et de Thérapie 152:25-58.
Yang Y, Wang Y, Li S, Xu Z, Li H, Ma L, Fan J, Bu D, Liu B, Fan Z, Wu G, Jin J, Ding B, Zhu X, Shen Y (2004).Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. Journal of Medical Genetics 41:171–174.
Cox JJ, Feimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM and Woods CG (2006) An SCN9A channelopathy causes congenital inability to experience pain. Nature 444:894-898.
Huang et al (2022) Structural basis for high-voltage activation and subtype-specific inhibition of human Nav1.8. Proc Natl Acad Sci U S A. 119:e2208211119.
Huaizong Shen, Dongliang Liu, Kun Wu, Jianlin Lei, Nieng Yan (2019) Structures of human Nav1.7 channel in complex with auxiliary subunits and animal toxins. Science, 363(6433):1303-1308.
Xiaojing Pan, Zhangqiang Li, Qiang Zhou, Huaizong Shen, Kun Wu, Xiaoshuang Huang, Jiaofeng Chen, Juanrong Zhang, Xuechen Zhu, Jianlin Lei, Wei Xiong, Haipeng Gong, Bailong Xiao, Nieng Yan (2018) Structure of the human voltage-gated sodium channel Nav1.4 in complex with β1. Science, 362(6412): eaau2486.
Huaizong Shen, Qiang Zhou, Xiaojing Pan, Zhangqiang Li, Jianping Wu, Nieng Yan (2017) Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science, 355(6328): eaal4326.
转自:饶毅 饶议科学 2023-08-10 07:30 发表于广东
痛觉不仅是人类与生俱有的生理、病理过程,也是可以上升到哲学层面讨论的问题。
如果能够有理想的镇痛药,可以随时随地抑制痛觉而没有其他副作用,可能人类的生活将有很大的变化。恐怕家家都会备有镇痛药,人人每天身上携带镇痛药。
但是,镇痛药的研究却非常不容易。虽然早就发现了鸦片的镇痛作用,但几千年来无法分开其镇痛作用与其成瘾作用。
寻找没有成瘾作用的镇痛药,是人类长期的愿望。
而镇痛药物的研究,又帮助科学理解痛觉的机理。
13.1 鸦片
鸦片来自罂粟(Papaver somniferum),最早历史记载是公元前3000到4000年,苏美尔人种植,罂粟汁制造鸦片(Blakemore and White,2002)。古希腊始称为鸦片,在荷马史诗《伊利亚特》和《奥德赛》中都提到了鸦片。

鸦片具有减少痛疼、催眠作用,也能够治疗一部分腹泻。当然,鸦片也能够导致欣快感和成瘾。鸦片的滥用损害人类健康,也造成了社会的悲剧。
鸦片曾是人类很重要的药物。
英国医学之父Thomas Sydenham(1624-1689)曾称鸦片为上苍给人类减缓病患、甚至治疗某些疾病最强的药物(Jones, 1700;Wright,1968)。

13.2 吗啡
1803年,法国药剂师Jean-François Derosne(1774-1855)首先报道从鸦片中提取活性成分(Derosne,1803;Holmes,1952)。
1804年,法国化学家Armand Séguin(1767-1835)宣读他与Bernard Courtois(1777-1838)合作提取鸦片化学成分的工作(Séguin,1814)。
德国药剂师Friedrich Sertürner(1783-1841)于1805和1806年发表其从鸦片中提取化学成分的结果(Sertürner,1805,1806),1817年再发表更详细的阐述,参照希腊梦神(morpheus)的名字而称为morphium。

翻译为法文时法国物理和化学家Joseph Louis Gay-Lussac (1778-1850)称之为“morphine”(Sertürner,1817)。

1831年,德国化学家Justus von Liebig(1803-1873)分析后提出吗啡的化学分子式(C34H36O6N2),于1847年被法国化学家Auguste Laurent(1807-1853)纠正为C17H19NO3(Laurent,1847)。
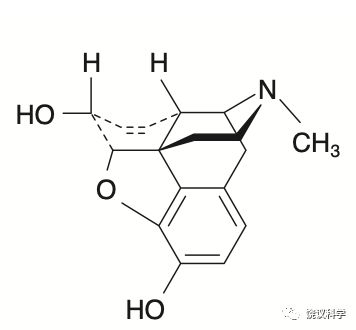
英国曼彻斯特大学的化学家John Gulland (1898-1947)和Robert Robinson (1886-1975)提出了吗啡的结构(Gulland and Robinson,1923,1925),立体化学构型在1950年代确定(Holmes and Stork,1952;Mackay and Hodgkin,1955;Bentley and Cardwell,1955)。美国Rochester大学化学系的Marshall Gates(1915-2003)和Gilg Tschudi人工全合成吗啡(Gates and Tschudi,1952,1956)。
在理解吗啡化学结构的基础上,人工合成多种衍生分子(Beckett,1952),其中一些失去活性。失去活性的与有活性的分子,有些差别很小:分子式一样、只是立体结构有差别的立体异构分子,可以一个有活性,另一无活性。有些衍生物与吗啡作用类似,为吗啡激动剂。有些衍生物本身不引起生理学反应但可以抑制吗啡的作用,为吗啡拮抗剂,如naloxone等(Kosterlitz and Watt,1968)。
13.3 痛觉传导通路中的特异基因
痛觉对于保护人类非常重要,没有痛觉的人难以长期健康生活。先天缺乏痛觉的人会出现认知偏差,难以活到成年。
曾经认为只要刺激到达伤害程度,外周任何神经纤维末梢被过度刺激都会向中枢传导痛觉信号。通过长期的研究证明,痛觉传导通路与其他体躯感觉如温度、触压、痒的传导通路是分开的,而不是其他感觉过度强烈后成为痛觉(Perl, 2007)。
现在认为,有特异的痛觉神经,专门传输痛觉信息。一般来说,伤害性刺激由于其物理、化学性质可以特异地激活痛觉神经末梢,由痛觉神经传导信息至脊髓,上传至脑,经过引起痛觉(Basbaum et al., 2009)。
世界上,真有缺乏痛觉的人,当然极为罕见。科学上报道第一例缺乏痛觉的人是1932年(Dearborn, 1932),迄今也不过十几例(Landrieu, Said and Allaire,1990;Nagasako, Oaklander and Dworkin,2003;Cox et al., 2006)。在巴基斯坦发现三个遗传痛觉缺失的家系,都是近亲结婚的家庭。其中一位小孩14岁不到就去世,原因是他没有痛觉,平时经常在街头表演刀刺自己、火上行走,他不感到痛,但这并不意味着他不受伤害,而是受了伤害不感到痛。人如果从来不感到痛,对世界的理解会出现偏差。这个孩子后来跳楼,当然他确实不痛,但与常人一样,跳楼可以导致死亡。
还有一种病,称为“红斑肢痛症”(erythermalgia)(Mitchell,1878),患者会自发或者受到轻微的刺激就出现肢体剧烈疼痛、皮肤红、灼热。1966年,美国Mayo医院的医生发现阿拉巴马州有一个较大的家系,5代51人中21位有自发疼痛(Burbank, Spittell and Fairbairn, 1966)。1992年,进一步发现这一家系有更多(共29)人患病(Finley et al., 1992)。荷兰遗传学家联系美国阿拉巴马州的医生,获得这一家系多个病人和正常人的DNA样本,进行分子遗传分析,发现病人是因为2号染色体特定段落出现突变,这段含约50个基因,但他们不知道是哪个基因导致自发疼痛。

北京大学第一医院皮肤科的杨勇,发现一家三代有“红斑肢痛症”。他进一步分析了这50个基因,发现包含有一组钠通道基因,而其中SCN9A比较特异地表达在外周神经,与伤害感受有关,是最可能的致病基因。他们发现SCN9A基因在一个3代13人家系中每一病人(共7位)都有相同的点突变,而正常人没有。散发病人有一位也在同一基因有另外一个点突变。杨勇因此提出SCN9A突变是“红斑肢痛症”的致病原因(Yang et al., 2004)。SCN9A基因这些点突变导致钠离子通道蛋白功能增强,从而感到痛(Cummins, Dib-Hajj and Waxman,2004)。
2006年,英国遗传学家专门研究巴基斯坦三个先天缺乏疼痛的家系,三家有六位完全没有痛觉。分析发现他们都是有SCN9A的功能缺失型突变而失去痛觉。他们的触觉、压觉、温度感觉、本体感觉没有受影响(Cox et al., 2006)。
13.4 吗啡的中枢镇痛作用
痛觉又经常让人们难受,人们经常希望能够有镇痛的方法。
镇痛的药物可以在外周发挥镇痛作用、也可能在中枢发挥镇痛作用。
1964年,当时在美国Miles药厂工作的林可胜(Robert KS Lim, 1897-1969)发明了区分药物是在中枢还是在外周起镇痛作用的方法(Lim et al., 1964)。林可胜的实验设计是:将一只狗(受体狗,R)的脾脏完全与其自身血流分开,而接受供体狗(D)的血流灌注。其他照常(也就是说,R的其他器官包括中枢神经系统继续由R的血流灌注,R的脾神经也原封不动)。已知通过血液给R的脾注射缓激肽会引起痛疼(通过狗叫、呼吸和血压监测)。在给D注射阿司匹林时,可以抑制痛觉,而给R注射阿司匹林时,不能抑制痛觉。这些实验说明阿司匹林的作用在脾脏(外周)发挥,而不是在中枢神经系统发挥。同样的实验,如果注射吗啡,只有注射到R才能抑制痛觉,而注射到D没有用,说明吗啡镇痛作用不在外周发挥,而可能在中枢发挥(Lim et al., 1964)。
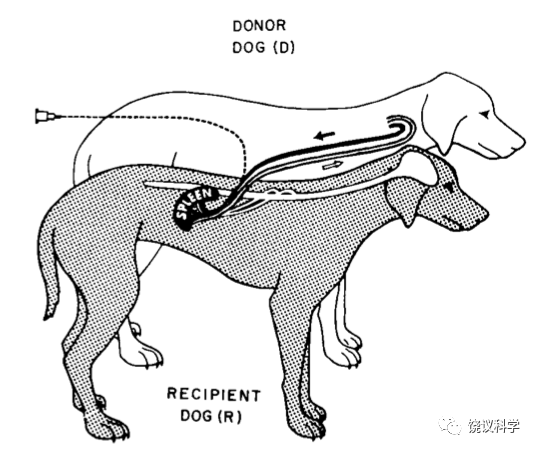
林可胜通过这一优美实验设计建立了鉴定中枢和外周镇痛的一个标准模型,可以鉴定很多药物的外周、中枢差别(Lim,1968)。林可胜还有两个实验证明阿司匹林的外周镇痛作用:脾脏注射致痛的缓激肽,引起脾感觉神经发放冲突增加,这一作用可以被阿司匹林所阻断,不能被吗啡所阻断;脾脏注射缓激肽引起痛觉,如果阿司匹林通过与缓激肽同样的局部途径注射进入脾脏,其镇痛所需要的剂量(3.8 mg/kg)低于静脉注射阿司匹林能够镇痛剂量(50 mg/kg)的10%,而八倍(30 mg/kg)的阿司匹林注射到头颈动脉也不能引起镇痛(Lim et al., 1964;Guzman et al., 1964)。与此相反,在头颈动脉注射吗啡引起镇痛所需剂量低于局部脾脏注射吗啡所需的剂量(Lim,1970)。
林可胜用三个实验证明阿司匹林镇痛作用的位点在外周,是阿司匹林研究的里程碑之一。阿司匹林是如何在外周发挥镇痛作用的呢?其生物化学机理是抑制前列腺素的合成(Vane, 1971; Smith and Willis, 1971),而有些种类的前列腺素可以刺激痛觉系统的外周神经末梢,因此阿司匹林能够缓解一部分疾病的痛疼。
林可胜的研究结果也支持了1962年中国科学家的发现:吗啡在中枢神经系统发挥镇痛作用。
13.5 吗啡镇痛位点
研究吗啡促进了对痛觉的理解。
吗啡镇痛的作用位点在脑内(Wikler, 1950;Lockett and Davis, 1958)。例如中国科学院药物研究所的周金煦和胥彬发现脑室内注射吗啡产生镇痛作用所需要的药量是皮下注射产生镇痛作用的百分之一,支持吗啡可能在脑内起镇痛作用(Chou and Hsu,1959)。吗啡镇痛作用的位点不是大脑皮层或下丘脑(Masserman, 1939)。但不清楚吗啡作用具体在脑内什么部位。
1962年,中科院药物所的研究生邹冈(1932-1999)与其导师张昌绍(1906-1967)在《生理学报》以中文全文和英文摘要发表其研究结果(Tsou and Jang,1962),1964年再以英文发表全文:“脑室内或脑组织内微量注射吗啡的镇痛效应” (Tsou and Jang,1964)。
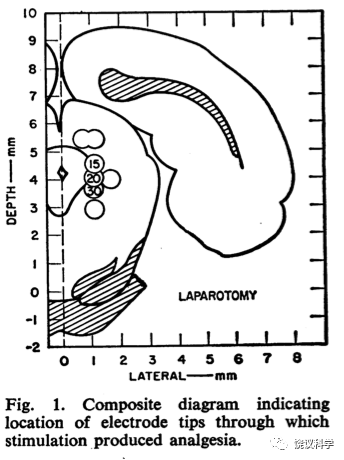
邹和张首先给家兔脑室内注射吗啡,可以观察到镇痛作用。比较注射到脑室和注射到静脉后的吗啡镇痛作用,发现注射到脑室产生镇痛作用所需要的吗啡剂量是注射到静脉所需剂量的1/500-1/1000,而镇痛作用时间也是脑室注射的更长。将脑室注射有效的剂量相同的吗啡放在滤纸上贴在大脑皮层没有镇痛作用。他们在皮层下多个部位注射微量吗啡,发现吗啡注射到第三脑室周围灰质有显著镇痛作用,而且可以被吗啡拮抗剂丙烯吗啡所抑制。

1969年,美国科学家发现电刺激大鼠中脑中央灰质可以镇痛(Reynolds,1969),这一发现说明脑内有内源镇痛机制,在电刺激激动下可以主动抑制痛觉。这一发现得到广泛验证:鼠(Mayer et al., 1971;Balagura and Ralph,1973;Melzack and Melinkoff,1974)、猫(Liebeskind et al.,1973;Oliveras et al.,1973)、猴(Goodman and Holcombe,1976)、人(Mayer DJ and Liebeskind,1974)。
电刺激引起镇痛的部位,与邹冈和张昌绍发现的吗啡镇痛部位相同:中脑导水管周围灰质(periaqueductal gray,PAG)(Mayer and Price,1976;Hayes et al.,1979)。而且,电刺激PAG的镇痛作用可以被吗啡拮抗剂所抑制(Adams,1976;Akil, Maycr and Liebeskind,1976);Hosobuchi, Adams and Linchitz,1977;Richardson and Akil,1977;Lewis and Gebhart,1977)。
由此猜测内源镇痛系统可能是类似吗啡的物质所介导。
13.6 阿片受体
按药理学的推论和立体异构活性的差别(Beckett,1952),阿片类的分子发挥作用的最简单解释是作用于靶细胞的特异受体(Beckett and Casy,1954)。

1971年,为了探寻阿片受体,斯坦福大学药理系Avram Goldstein(1919-2012)发明了检测脑内阿片受体的方法。他们设计:放射性同位素标记阿片类分子,利用阿片类分子的立体特异性(一般D(-)构型的有作用,L(+)构型的同分异构体无作用),在将放射性有效阿片类分子与脑组织细胞结合的时候,加百倍的无标记的D(-)构型的同一分子,应该可以竞争放射性同位素标记的药物与细胞上特异受体的结合,而如果加百倍的无标记的L(+)构型的同一分子,应该不能竞争放射性同位素标记的药物与细胞上特异受体的结合,而能够减少其他非特异的结合,这样就能发现特异结合的受体(Goldstein,Lowney and Pal,1971)。这一方法行之有效,但Goldstein等最初用的标记的阿片类分子被同位素标记的比率较低,实验结果不好。
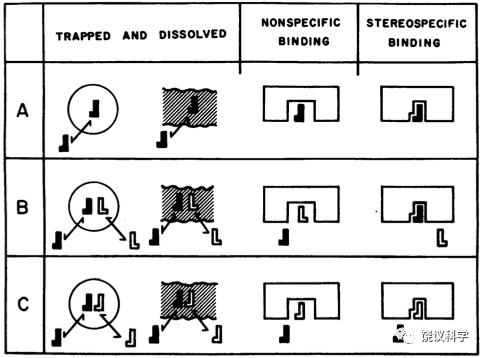
1973年,三个实验室用类似方法,用同位素标记效率更高的阿片类分子检测到特异的阿片受体。瑞典Uppsala大学的Lars Terenius(1940-)用氚标记的双氢吗啡(3H DHM)与来自脑的突触体膜制备进行结合实验,事先可以给老鼠多天注射无同位素标记的左旋或右旋吗啡拮抗剂methadone,发现可以被有活性的左旋methadone所竞争的结合(Terenius,1973a)。同一实验,可以直接在结合实验时加立体异构的阿片类分子,可以观察到立体异构特异的竞争性抑制,也可以观察到非阿片类分子不能特异地竞争抑制DHM与突触体膜制备的结合(Terenius,1973b)。

美国霍普金斯大学医学院的Candace Pert(1946-2013)和Solomon Snyder(1938-)用氚标记的吗啡拮抗剂纳洛酮与大鼠、小鼠或豚鼠的脑匀浆做结合实验,可以被D(-)构型的有活性的阿片类分子(如levorphanol)所抑制、不能被L(+)构型的无活性的阿片类分子(如dextrorphan)所抑制,也不能被胆碱、去甲肾上腺素、组织胺、五羟色胺等所抑制(Pert and Snyder,1973)。他们还发现,如果比较氚标纳洛酮与豚鼠小肠或与脑的结合,levorphanol抑制脑组织与氚标纳洛酮结合的强度40倍于其抑制小肠与氚标纳洛酮的结合,levorphanol抑制脑组织与氚标纳洛酮结合的强度500倍于其抑制小肠与氚标纳洛酮的结合。而在脑与氚标纳洛酮结合实验中,levorphanol与dextrorphan差别有4000倍,而在小肠与氚标纳洛酮结合实验中,levorphanol与dextrorphan差别仅500倍。这些结果提示脑内和小肠的阿片受体有所不同。不同脑区,阿片受体的含量不同(Pert and Snyder,1973)。

美国纽约大学的犹太裔药理学家Eric Simon(1924-2020)等用氚标记当时作用最强的吗啡样分子etorphine,可以与大鼠脑匀浆进行可饱和的、可以被立体异构特异的分子所抑制,抑制强度基本与激动剂、拮抗剂强度相关(Simon, Hiller and Edelman,1973)。
1971至1973年的研究证明:脑内存在特异的与吗啡类分子结合的物质,很可能是介导吗啡药理作用的受体。
13.7 内源性阿片样物质
1960年,美国威斯康辛大学的科学家发现垂体后叶含增强吗啡样物质镇痛作用的活性(Murray and Miller,1960)。

德国犹太裔科学家Hans Kosterlitz(1903-1996)于1934年不得不离开柏林Charité医院,移民苏格兰加入Aberdeen大学生理系,1968年他创立其药理系并任第一任系主任。1973年退休后,他继续有实验室,研究成瘾药物。早先,他和一些药理学家就思考过身体多个组织对吗啡有反应提示可能存在内源性吗啡样物质,而1973年阿片受体被证明,更提示受体不应该是为植物来源的生物碱吗啡所存在,而应该有内源性配体。他与药理系讲师John Hughes讨论分离纯化内源性阿片样物质:用灵敏的检测方式、用拮抗剂保证特异性(Kosterlitz,1979)。

Hughes和Kosterlitz没有用放射性同位素的受体结合实验,而是用传统的生物检定(bioassays):吗啡可以引起豚鼠回肠收缩(Trendelenburg,1917;Schaumann,1955;Kosterlitz and Robinson,1955),抑制刺激神经引起的猫的瞬膜平滑肌收缩(Trendelenburg,1957),抑制肾上腺素能神经兴奋引起的小鼠输精管收缩(Henderson, Hughes and Kosterlitz,1972;Hughes, Kosterlitz and Leslie,1975)。这些作用见效比放射性标记的配体结合实验要更快,Hughes在兔、豚鼠、大鼠、猪脑检测到有吗啡样生物活性的分子量小于700道尔顿的分子,其作用被纳洛酮等三种阿片受体拮抗剂所抑制,可以被多肽水解酶所降解(Hughes,1975)。
瑞典Uppsala大学的Terenius和Wahlström用他们1973年建立的氚标双氢吗啡与大鼠脑突触细胞膜结合实验检测,发现脑内有分子量在1000到1200道尔顿的肽链物质可以竞争氚标双氢吗啡结合脑匀浆(Terenius and Wahlström,1975)。
1975年5月24日,三个课题组同时在同期《生命科学》杂志上发表4篇文章,报道他们纯化内源性阿片样物质的进展。Hughes等从猪脑纯化到一个分子量在1000至1200道尔顿的多肽,在小鼠输精管上具有吗啡样的活性,其活性可以被阿片受体拮抗剂所阻断,而且依赖拮抗剂的立体异构。他们将之命名为脑啡肽(encephalin),其分布并不限于中枢神经系统,而与阿片受体分布相关,所有也应该与吗啡一样具有镇痛和其他中枢神经系统作用之外的作用(Hughes et al.,1975)。用竞争3H纳洛酮和3H双氢吗啡结合脑匀浆为检测方法,Pasternak、Goodman和Snyder 从大鼠和小牛脑分离到“吗啡样物质”(MLF),其分布与阿片受体高度相关(例如纹状体的阿片受体最多,MLF也最多),存在于突触体亚细胞制备中,可以被多肽酶降解,分子量约1000道尔顿(Pasternak, Goodman and Snyder,1975)。Goldstein实验室从猪的垂体分离到具有阿片活性的物质,在豚鼠肠道肌肉和小鼠输精管有阿片样活性,也能够竞争性抑制3H etorphine与脑组织的结合,其作用可以被立体异构特异的阿片受体拮抗剂所阻断(Teschemacher et al.,1975)。因为移植垂体含多种生物活性肽类分子,他们进一步检测已知多肽是否有吗啡有活性,ACTH的粗提物有活性,但合成的没有活性,其他多肽也没有吗啡样活性,他们进一步分析认为ACTH的粗提物和垂体含另外一个肽类分子有吗啡样活性,分子量1750道尔顿(Coxet al.,1975)。
1975年12月12日,Terenius报道他分析已知肽类分子时,发现促肾上腺皮质激素(ACTH)的1-28和4-10肽段可以竞争性抑制氚标双氢吗啡结合突触细胞膜(Terenius,1975)。
1975年12月18日,Hughes和Kosterlitz等报道他们分离纯化到的脑啡肽的氨基酸序列,甲硫氨酸脑啡肽(met-enkephalin)为Tyr-Gly-Gly-Phe-Met、亮氨酸脑啡肽(leu-enkephalin)为Tyr-Gly-Gly-Phe-Leu。在小鼠输精管的生物检定,两者都具有类似吗啡的作用(Hughes et al.,1975)。美国的Snyder实验室在1976年7月报道他们用纳洛酮结合竞争的方法,从牛脑也纯化到同样这两个脑啡肽 (Simantov and Snyder,1976)。
1976年,三个课题组报道他们分离纯化到内啡肽(b-endorphin)(Guillemin,Ling and Burgus,1976;Bradbury et al.,1976;Li and Chung,1976;Li et al.,1976),是李卓皓1965年从垂体分离多肽的一部分(Li et al.,1965)。

Goldstein于1975年发现的活性(Cox et al.,1975),经过几年的努力,终于1979年分离纯化到一个13肽,他命名为强啡肽(dynorphin),因为在豚鼠回肠纵肌的活性为脑啡肽的700倍(Goldstein et al.,1979)。

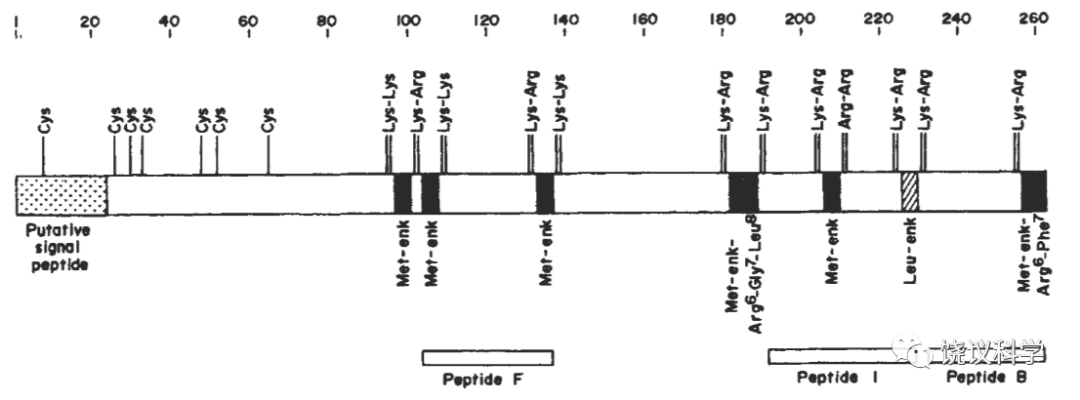
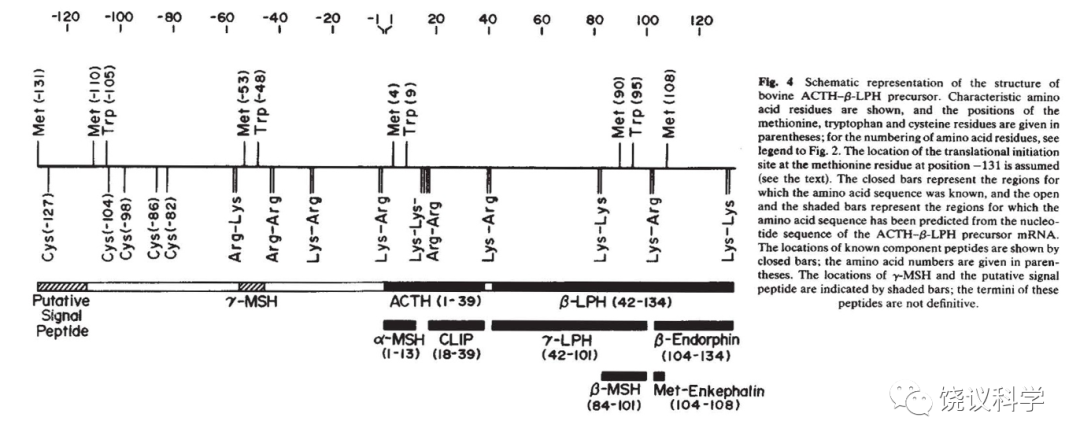
日本京都大学的沼正作(Shosaku Numa,1929-1992)通过克隆和分析cDNA序列,推出内啡肽、脑啡肽、强啡肽的前体蛋白质序列(Nakanishi et al.,1979;Noda et al.,1982;Kakidani et al.,1982)。他们都具有氨基端的信号肽(亦可称pre区),可以分泌,后接对加工需要的pro区,后面再是成熟的部分,而其中含成对出现的碱性氨基酸(K或R),为特定蛋白酶水解,从而产生小肽。例如:ACTH前体可以产生一个beta内啡肽(Nakanishi et al.,1979),脑啡肽前体可以产生6个甲硫氨酸脑啡肽、一个亮氨酸脑啡肽(Noda et al.,1982),强啡肽前体可以产生一个强啡肽、3个亮氨酸脑啡肽和一个b新内啡肽(Kakidani et al.,1982)。
13.8 镇痛药机理研究的意义
化学生物学最佳例子是吗啡。因为对吗啡的研究,发现了内源性镇痛的分子和神经机理。
内啡肽是神经活性多肽的一种。
第一个神经肽是P物质,它于1931年为von Euler和Gaddum发现(Euler and Gaddum,1931),到1971年其氨基酸序列为Leeman实验室所确定(Chang and Leeman,1970,1971)。

神经肽还有催产素(oxytocin,OXT)和抗利尿激素(arginine vasopressin,AVP),血管活性肠肽(VIP),神经肽Y(NPY)等。在中枢神经系统内,九肽OXT和AVP对社会行为与认知很重要,而它们在外周的作用还有其他作用。五十多个神经肽通过其受体在体内发挥广泛的作用。
并非外源化学分子对人体有作用就一定对应人体内源配体。化学分子可以作用于其他靶点,并不一定这些靶点都有类似药物的内源性物质。可以利用作用于受体的外源药物来预计内源配体。进一步找到内源性物质推动理解生物学机理,但不一定是一蹴而就。发现内源性阿片样物质,对痛觉理解很重要,但我们迄今对痛觉理解还有限,我们更不能分开吗啡的镇痛和成瘾作用。生物学对研究者的吸引力之一在于难以穷尽,激发研究者不断努力。
注1:德国化学家 Friedrich Gren (1760-1798)于1791 年提出药物学(materia medica) 和药理学(pharmacology)的区别:描述和收集药物为药物学,研究药物作用的科学为药理学。法兰西学院生理学家François Magendie (1783-1855)从1831年起先在动物做药物实验,从动物得到能够预计效果之后再在人身上做试验,以生理学途径建立了实验药理学。他的学生Claude Bernard (1813–1878)对美洲筒箭毒作用和一氧化碳结合血红蛋白的研究也同样从生理途径研究药理学。这样不仅研究药物作用机理,也推动生理学原理研究。德国科学家Rudolf Buchheim (1820–1879)在药物学的基础上,建立了实验室,1865 年正式命名为药理学研究所,成为世界上第一个独立的药理学研究所,他培养 了九十多个学生(Habermann,1974;Jansone et al. ,2016)。他的学生 Oswald Schmiedeberg (1838-1921)培养了来自二十多个国家一百二十多位学生,让药理学传 遍世界。他的美国学生John Jacob Abel (1857-1938)留学德国7年,1891 年成为美国第一位药理学教授、1893 年成为美国第一位药理系主任,创办了《生物化学杂志》 (JBC)和《药理学与实验治疗学杂志》(JPET)。Schmiedeberg 的另一学生 Hans Horst Meyer (1853-1939)最著名的工作是提出全身麻醉药的药效与其脂溶性相关,可能是因为全麻药融在细胞脂膜而干扰神经细胞的功能。
注2:1955年,Goldstein创办斯坦福大学医学院药理系,1965年创办Molecular Pharmacology杂志。
注3: Hughes、Kosterlitz和Snyder获1978年Lasker奖。估计是领域争议(特别是Pert和Snyder之间)导致了诺奖的犹豫。Kosterlitz的儿子、英裔美国物理学家Michael Kosterlitz于2016年获物理诺奖。

参考文献
Adams JE (1976) Naloxone reversal of analgesia produced by brain stimulation in the human. Pain 2:161-166.
AkilH, Maycr DJ and Liebeskind JC (1976) Antagonism of stimulation-produced analgesia by naloxone, a narcotic antagonist. Science 191:961-962.
Balagura S and Ralph T (1973) The analgesic effect of electrical stimulation of the diencephalon and mesencephalon. Brain Research 60:369-381.
Basbaum AI, Bautista DM, Scherrer G and Julius D (2009) Cellular and molecular mechanisms of pain. Cell 139:267-284.
Beckett AH (1952) Analgesics-a general survey. Journal of Pharmacy and Pharmacology 4:425-447.
Beckett AH and Casy AF (1954) Synthetic analgesics: stereochemical considerations. Journal of Pharmacy and Pharmacology 6:986-1001.
Beckett AH, Casy AF and Harper (1956) Analgesics and their antagonists: some steric and chemical considerations: Part III. The influence of the basic group on the biological response. Journal of Pharmacy and Pharmacology 8:874-884.
Bentley KW and Cardwell HME (1955) The morphine-Thebaine group of alkaloids. Part V. The absolute stereochemistry of the morphine, benzylisoquinoline, aporphine, and tetrahydroberberine alkaloids. Journal of Chemical Society 3252-3260.
Blakemore PR and White JD (2002) Morphine, the Proteus of organic molecules. Chemical Communications 83:1159-1168.
Bradbury AF, Smyth DG, Snell CR, Birdsall NJM and Hulme EC (1976) C fragment of lipotropin has a high affinity for brain opiate receptors. Nature 260:793-795.
Burbank MK, Spittell JA Jr., Fairbairn JF II (1966) Familial erythromelglgia: genetic and physiologic observations. Journal of Laboratory and Clinical Medicine 68:861.
Chang MM and Leeman SE (1970) Isolation of a sialogogic peptide from bovine hypothalamic tissue and its characterization as substance P. Journal of Biological Chemistry 245:4784-4790.
Chang MM, Leeman SE and Niall HD (1971) Amino-acid sequence of substance P. Nature New Biology 232:86-87.
Chou CH and Hsu B (1959) The analgesic action of and the tolerance to morphine give intracerebrally in mice. Acta Physiologica Sinica 23:37-44.
Corbett AD, Paterson SJ, McKnight AT, Magnan J andKosterlitz HW (1982) Dynorphin and dynorphin are ligands for the kappa-subtype of opiate receptor. Nature 299:79-81.
Cox BM,Opheim KE, Teschemacher H and Goldstein A (1975) A peptide-like substance from pituitary that acts like morphine. 2. Purification and properties. Life Sciences 16:1777-1782.
Cox JJ, Feimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM and Woods CG (2006) AnSCN9A channelopathy causes congenital inability to experience pain. Nature 444:894-898.
Cummins TR, Dib-Hajj SD and Waxman SG (2004) Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. Journal of Neuroscience 24:8232-8236.
Dearborn G (1932) A case of congenital general pure analgesia. Journal of Nervous and Mental Diseases 75:612-615.
Derosne CL (1803)Mémoires sur l’opium. Annales de chimie 45:257-285.
Euler USv and Gaddum JH (1931) An unidentified depressor substance in certain tissue extracts. Journal of Physiology72:74-87.
Finley WH, Lindsey JR Jr, Fine J-D, Dixon GA and Burbank MK (1992) Autosomal dominant erythromelalgia. American Journal of Medical Genetics 42:310-315.
Gates M and Tschudi G (1952) The synthesis of morphine. Journal of the American Chemical Society 74:1109-1110.
Gates M and Tschudi G (1956) The synthesis of morphine. Journal of the American Chemical Society 78:1380-1393.
Goldstein A, Lowney LI and Pal BK (1971) Stereospecific and nonspecific interactions of the morphine congener levorphanol in subcellular fractions of mouse brain. Proceedings of the National Academy of Sciences USA 68:1742-1747.
Goldstein A, Tachibana S, Lowney LI, Hunkapiller M and Hood L (1979) Dynorphin-(1–13), an extraordinarily potent opioid peptide. Proceedings of the National Academy of Sciences USA 76:6666–6670.
Goldstein A, Fischli W, Lowney LI, Hunkapiller M and Hood L (1981) Porcine pituitary dynorphin: complete amino acid sequence of the biologically active heptadecapeptide. Proceedings of the National Academy of Sciences USA 78:7219-23.
Goodman SJ and Holcombe V (1976) Selective and prolonged analgesia in monkey resulting from brain stimulation. In: Advances in Pain Research and Therapy, edited by Bonica JJ and Albe-Fesard D, New York: Raven, 1:495-502.
Guillemin R, Ling N and Burgus R (1976) Endorphines, peptides, d’origine hypothalamique et neurohypophysaire à activité morphinomimétique. Isolement et structure moléculaire de l’a-endorphine. Comptes Rendus de l’Académie des Sciences Paris 282:783-785.
Gulland JM and Robinson R (1923) The morphine group. Part I. A discussion of the constitutional problem. Journal of Chemical Society 123:980-998.
Gulland JM and Robinson R (1925) Constitution of codeine and thebaine. Memoirs and Proceedings of the Literary and Philosophical Society of Manchester 69:79-86.
Guzman F, Braun C, Lim RK, Potter GD and Rodgers DW (1964) Narcotic and non-narcotic analgesics which block visceral pain evoked by intra-arterial injection of bradykinin and other analgesic agents. Archives Internationales de Pharmacodynamie et de Thérapie 149:571-588.
Habermann ER (1974) Rudolf Buchheim and the beginning of pharmacology as a science. Annual Review of Pharmacology 14:1-9.
Hayes RL, Price DD Ruda M and Dubner R (1979) Suppression of nociceptive responses in the primate by electrical stimulation of the brain or morphine administration: Behavioral and electrophysiological comparisons. Brain Res167:417-421.
Henderson G,Hughes J and Kosterlitz HW (1972) A new example of a morphine-sensitive neuro-effector junction: adrenergic transmission in the mouse vas deferens. British Journal of Pharmacology 46:764-766.
Hosobuchi Y, Adams JE and Linchitz R (1977) Pain relief by electrical stimulation of the central gray matter in humans and its reversal by naloxone. Science 197:183-186.
HolmesHL (1952) The morphine alkaloids. I. The Alkaloids: Chemistry and Physiology 2:1-159 Vol III, Chapter 8, Part 1. Academic Press, London.
Holmes HL and Stork G (1952) The morphine alkaloids. In: Manske RHF, Holmes HL (eds) The alkaloids, Vol III, Chapter 8, Part 2. Academic Press, London.
Hughes J (1975) Isolation of an endogenous compound from the brain with pharmacological properties similar to morphine. Brain Research 88:295-308.
Hughes J, Smith T, Morgan B and Fothergill L (1975) Purification and properties of enkaphalin-the possible endogenous ligand for the morphine receptor. Life Sciences 16:1753-1758.
Hughes J, Smith TW, Kosterlitz HW, Fothergill LA, Morgan BA and Morris HR (1975) Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature 258:577-580.
Hughes J,Kosterlitz HW and Leslie FM (1975) Effect of morphine on adrenergic transmission in the mouse vas deferens. Assessment of agonist and antagonist potencies of narcotic analgesics. British Journal of Pharmacology 53:371-381.
Jansone B, Kuum M and Maciulaitis R (2016) Pharmacology in Estonia, Latvia and Lithuania: from historical roots to nowadays achievements. Pharmacological Research 113:723-730.
Jones J (1700) The mysteries of opium reveal’d. Richard Smith, Angel and Bible, London.
Kakidani H, Furutani Y, Takahashi H, Noda M, Morimoto Y, Hirose T, Asai M, Inayama S, Nakanishi S and Numa S (1982) Cloning and sequence analysis of cDNA for porcineβ-neo-endorphin/dynorphin precursor. Nature 298:245-249.
Kosterlitz HW (1979) The best laid schemes o’ mice an’ men gang aft agley. Annual Review of Pharmacology and Toxicology 19:1-12.
Kosterlitz HW and Robinson JA (1955) Mechanism of the contraction of the longitudinal muscle of the isolated guinea-pig ileum, caused by raising the pressure in the lumen. Journal of Physiology 129:18-19P.
Kosterlitz HW and Wallis DI (1966) The effects of hexamethonium and morphine on transmission in the superior cervical ganglion of the rabbit. British Journal of Pharmacology 26:334-344.
Kosterlitz HW and Waterfield AA (1975) In vitromodels in the study of structure–activity relationships of narcotic analgesics. Annual Review of Pharmacology 15:29-47.
Kosterlitz HW andWatt AJ (1968) Kinetic parameters of narcotic agonists and antagonists, with particular reference to N-allylnoroxymorphone (naloxone). British Journal of Pharmacology and Chemotherapeutics 33:266–276.
Landrieu P, Said G and Allaire C (1990) Dominantly transmitted congenital indifference to pain. Annals of Neurology 27:574-578.
LewisVA and GebhartG F (1977) Evaluation of the periaqueductal central gray (PAG) as a morphine-specific locus of action and examination of morphine-induced and stimulation-produced analgesia at coincident PAG loci. Brain Research 124:283-303.
Li CH,Barnafi L Chrétien M and Chung D (1965) Isolation and amino-acid sequence of b-LPH from sheep pituitary glands. Nature 208:1093-1094.
Li CH and Chung D (1976) Isolation and structure of an untriakontapeptide with opiate activity from camel pituitary glands. Proceedings of the National Academy of Sciences USA 73:1145-1148.
Li CH, Lemaire S, Yamashiro D and Doneen BA (1976) The synthesis and opiate activity ofb-endophin. Biochemical and Biophysical Research Communications 71:19-25.
Liebeskind JC, Guilbaud G, Besson JM and Oliveras J-L (1973) Analgesia from electrical stimulation of the periaqueductal gray matter in the cat: behavioral observations and inhibitory effects on spinal cord interneurons. Brain Research 50:441-446.
Lim RK (1968) Neuropharmacology of pain and analgesia. In Lim RKS, Armstrong D and Pardo EG (eds): Pharmacology of Pain, p169-217. Oxford: Pergamon Press.
Lim RK(1970) Pain. Annual Review of Physiology 32:269-88.
Lim RK, Guzman F, Rodgers DW, Goto K, Braun C, Dickerson GD and Engle RJ (1964) Site of action of narcotic and non-narcotic analgesics determined by blocking bradykinin-evoked visceral pain. Archives Internationales de Pharmacodynamie et de Thérapie 152:25-58.
Lockett MF and Davis MM (1958) The analgesic action of normorphine administrated intracisternally to mice. Journal of Pharmacy and Pharmacology 10:80-85.
Lord JA,Waterfield AA, Hughes J and Kosterlitz HW (1977) Endogenous opioid peptides: multiple agonists and receptors. Nature 267:495-499.
Mackay M and Hodgkin DC (1955) A crystallographic examination of th
Copyright© 上海博璞诺科技发展有限公司 2016-2019 沪ICP备16015171号 中国化工网 全球化工网 生意宝 著作权声明
地址:上海市浦东新区张江高科技园区海科路100号9B楼四楼 电话:+86-21-20608178 传真:+86-21-20608171 联系我们

