详细内容
转自: 段思研 GPCR drug discovery 2023-08-02 15:49 发表于北京
背景
阿片类镇痛剂是一类强效的镇痛药物,在全球范围内有着广阔的市场。由于它们的镇痛效果显著,被广泛用于术后疼痛、严重创伤、癌症患者以及其他慢性疼痛病例,这使得阿片类镇痛剂成为医疗领域中不可或缺的一部分。
然而,持续流行的阿片类药物仍然是影响全球社会和经济福利的严重公共卫生危机,潜在的问题包括上瘾风险、过量使用和中毒、耐受性和副作用规、滥用问题等。
开发治疗阿片类药物使用障碍 (opioid use disorders,OUD) 的创新药物以及安全、有效、无成瘾性的疼痛管理策略,同时最大限度地降低复发风险,是新型止痛剂研发的重点。
7月19日,来自美国 NIH 的 Amy Hauck Newman 教授在《药物化学》杂志撰文,设计并合成了具有优化理化性质的新一代双靶点 μ阿片受体(MOR)激动剂/多巴胺 D3 受体(D3R)拮抗剂。结合基于体外细胞水平的亲和力筛选、计算机辅助药物设计和 BRET 功能测试等,研究人员确定了新的结构支架,它们分别对 MOR 和 D3R 具有高亲和力和拮抗活性,提高了多巴胺受体亚型的选择性(例如 D3R 优于 D2R),并显著提高了预测血脑屏障通透性的中枢神经系统多参数优化分数。

思路
经过背景调研,作者计划选用多巴胺受体家族的成员 D3R,因为 D3R 主要表达于大脑中叶 DA 区域,该区域控制着与药物相关的线索、强化、动机和奖励相关的行为。许多实验室率先设计和临床前开发了高选择性 D3R 拮抗剂,作为治疗精神刺激剂使用障碍和 OUD 的药物疗法。
作者列举了2个候选药物 VK4-116 和 VK4-40等,它们不仅能在不降低抗痛觉的情况下降低 oxycodone (一种半合成的阿片类止痛药) 的药物寻求和自我给药,而且,单独给药或与 oxycodone 或可卡因同时给药时不会影响外周生物心血管参数。
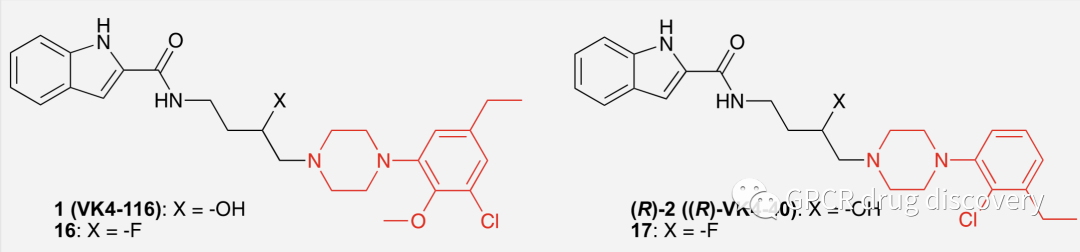
图片红色为标注的 D3R拮抗剂 的关键药效团
作者从已知的 μ-阿片受体的已报道安全激动剂入手,列举了4个近期研究较多的激动剂,包括 TRV130 (3)、PZM21、TRV734 以及 Loperamide (6),并选择其关键的药效团,用于下一步的双重配体开发。
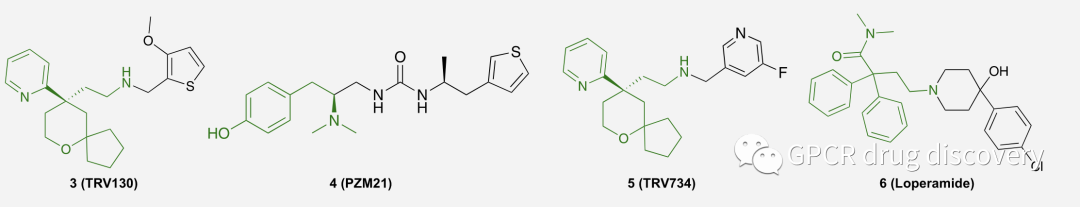
图片绿色为标注的 μ-阿片受体激动剂的关键药效团
策略:
作者将 μ-阿片受体激动剂的镇痛特性与 D3R 拮抗剂的降低成瘾性相结合,采用二价药物设计和支架杂交策略设计了化合物,将两种药理作用结合在一起:(i) μ-阿片受体激动剂药效团,靶向并激活 μ-阿片受体的正构配体口袋;(ii) D3R 拮抗剂药效团,在 D3R 正构配体口袋内结合。
作者通过具有不同取代、化学性质和立体化学模式(蓝色)的连接体连接 μ-阿片受体药效团(绿色)和 D3R 药效团(红色)PP,设计出新的双靶点药物库。
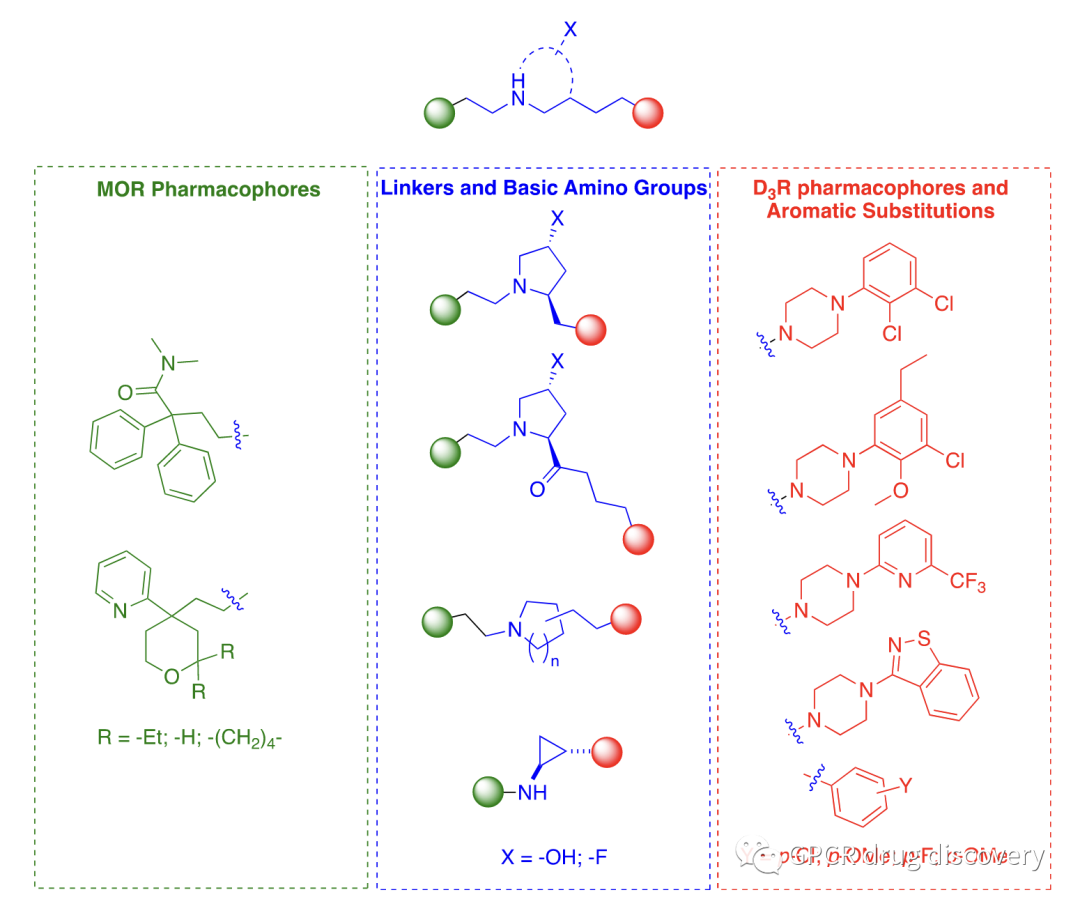
作者探索了 μ-阿片受体激动剂支架周围的取代模式、连接物的长度、立体化学和结构组成的变化,以及具有拮抗剂或部分激动剂功能的 D3R 药性等。
作者开发的双价配体可根据 μ-阿片受体激动剂药效团可分成两大类:
(A) 源自 6 (Loperamide) 的 N,N-二甲基-2,2-二苯基乙酰胺类;
(B) 源自 3 (TRV130) 及其类似物启发的 2-(四氢-2H-吡喃-4-基)吡啶类。
此外,本研究中,作者利用计算机辅助的计算方法 (中枢神经系统多参数优化分数CNS-MPO,计算药物穿过血脑屏障可能性的数值) 来预测新药靶向中枢神经系统的能力,并通过专门调整必要的理化性质来获得生成主要作用于外周的配体新系列,挖掘其在外周疼痛和炎症方面的治疗潜力。
方法和结果:
药物设计和合成
作者对所设计的分子,利用6个参数预测血脑屏障渗透性和中枢神经系统活性,包括分子量(MW)、clogP、clogD、pKa、拓扑极性表面积(TPSA)和氢键供体基团的数量。
为了满足双靶点 D3R-MOR 二价药效团需求,作者选择高分子量化合物、较高的clogP 值/clogD 值、偏碱的 pKa,这对各自的靶点结合至关重要。作者采用了以下设计来改进 CNS-MPO 计算中涉及的各项参数:
(i) 降低 MW、clog P 和 clog D (较大的clogP 值可提高跨膜渗透率,但同时,大的亲脂性分子也会表现出非特异性蛋白结合);
(ii) 降低碱性氮的 pKa 值
(iii)通过替换 H 键供体原子或加入能使 H 键供体参与分子内 H 键的 H 键受体来减少 H 键供体,这样做会降低水溶剂和 P-gp 的可用性,也会降低分子的灵活性
(iv)虽然 TPSA 并未被特别用作设计的一个因素,但去除杂原子会降低 TPSA 值,而加入 H 键受体则会提高该值。
以下为代表性的分子及理化参数:
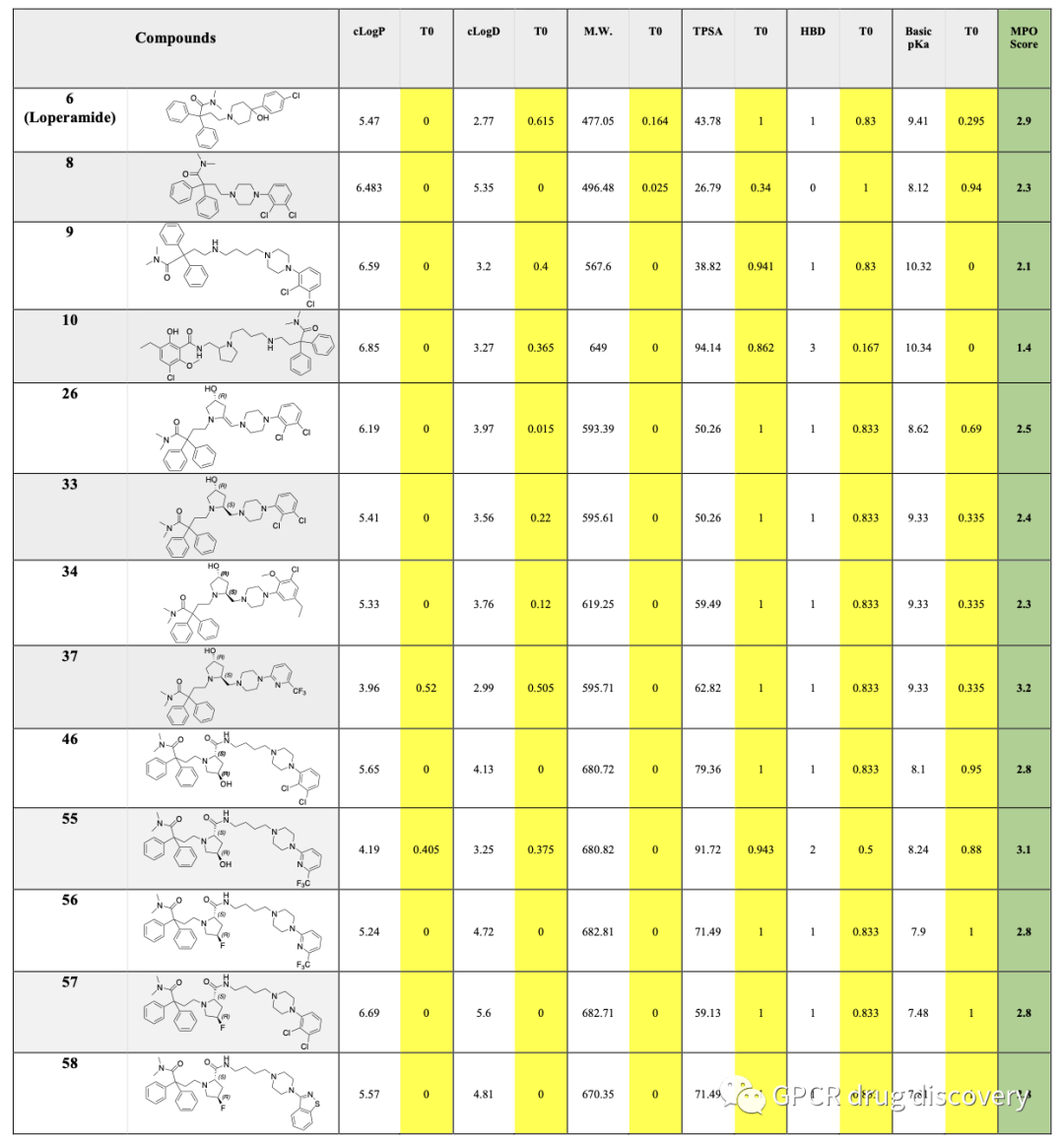
SARs 和CNS-MPO 分析
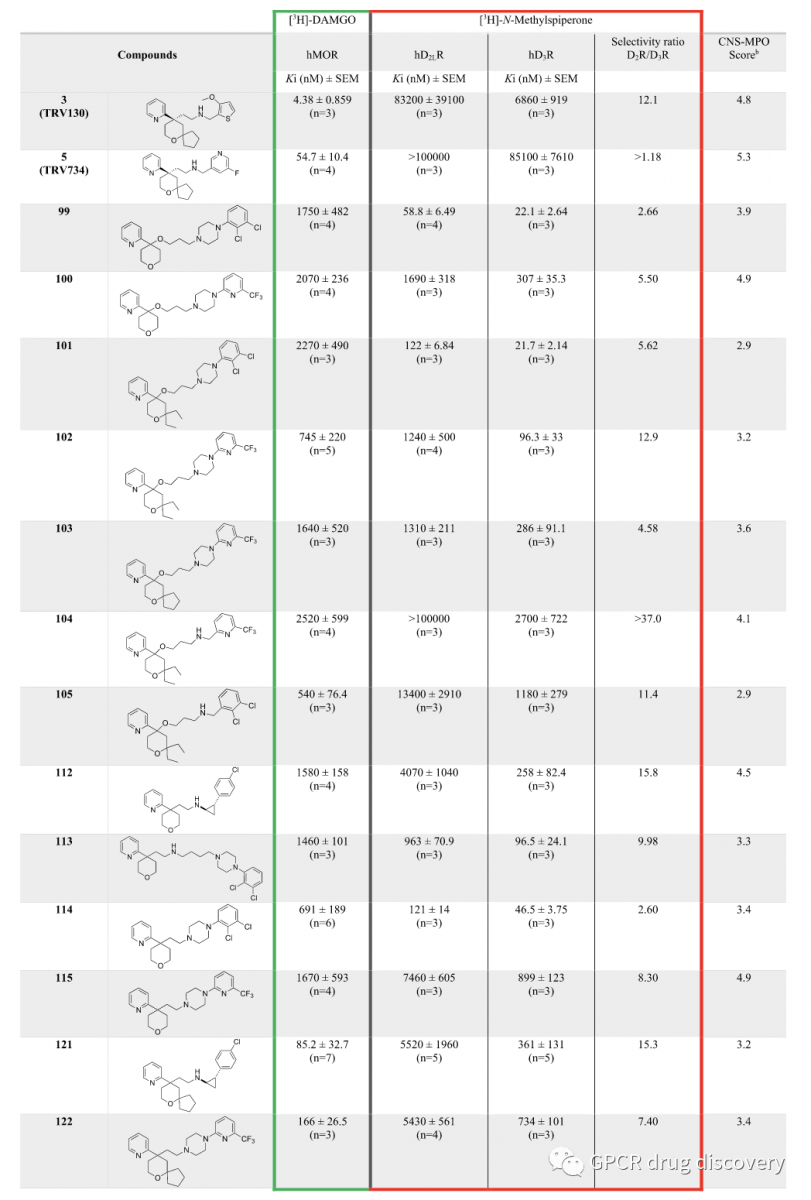
使用稳定表达的 HEK293 细胞系,分别与 [3H]-DAMGO (μ-阿片受体激动剂) 和 [3H]-N-methylspiperone (多巴胺受体拮抗剂) 竞争,评估了所有新类似物与 hMOR、hD2LR 和 hD3R 的结合亲和力 (Ki)及亚型选择性。
部分代表性分子的亲和多巴胺受体亚型选择性情况如下:
利用 BRET 方法进行μ-阿片受体和多巴胺受体的功能研究
根据化合物的结构、结合亲和力和 CNS-MPO 评分,作者选择了 13 个化合物进行体外功能研究 (33、34、46、55、56、58、 68、78、80 、 84、102、114 和 121),分别通过一系列 BRET 分析法,评估配体对G蛋白及 βarrestin 通路的激动活性,对无激动活性的分子,进一步进行拮抗属性鉴定。
BRET 系列的方法包括,针对 MOR 受体方面使用的构象选择性纳米抗体(Nb33)、arrestin-3 招募和 Gαi2 招募;在 D3R 方面,使用 GαoA 招募以及GαoA 的拮抗活性测定。
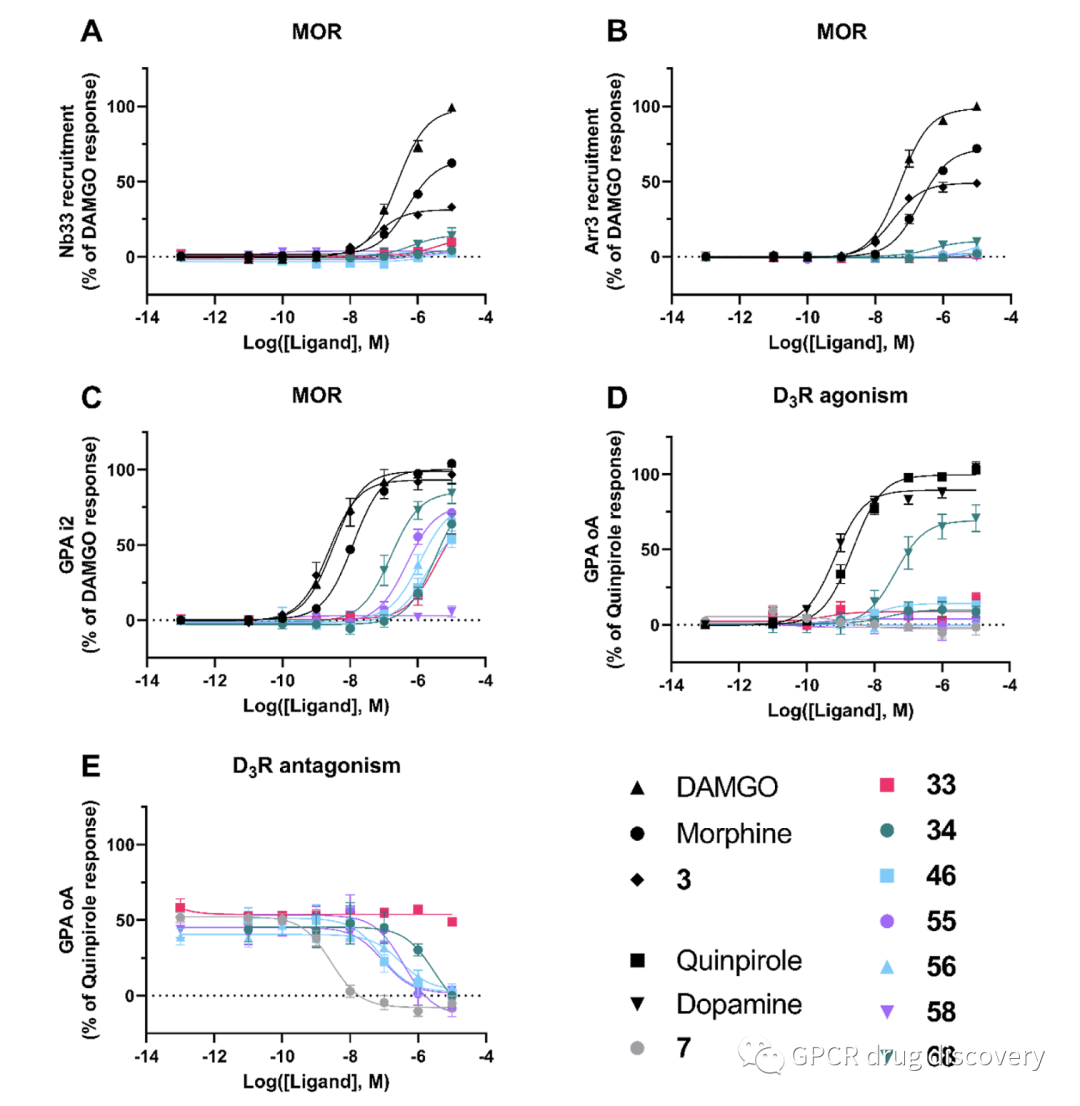
该系列的分子中,除58外,所有分子均表现出一定的激动μ-阿片受体的活性 (Gi),而几乎无激动Arr3活性;除68表现出部分激动剂 D3R 活性外,其余分子较弱的激动或具有拮抗活性,其中46 具有较强的 D3R 拮抗剂性质。
该系列的分子中,78、80、84均表现出完全激动μ-阿片受体的活性 (Gi),部分激动Arr3活性;所有分子均无 D3R 激动活性,其中80和84 具有较强的 D3R 拮抗剂性质。
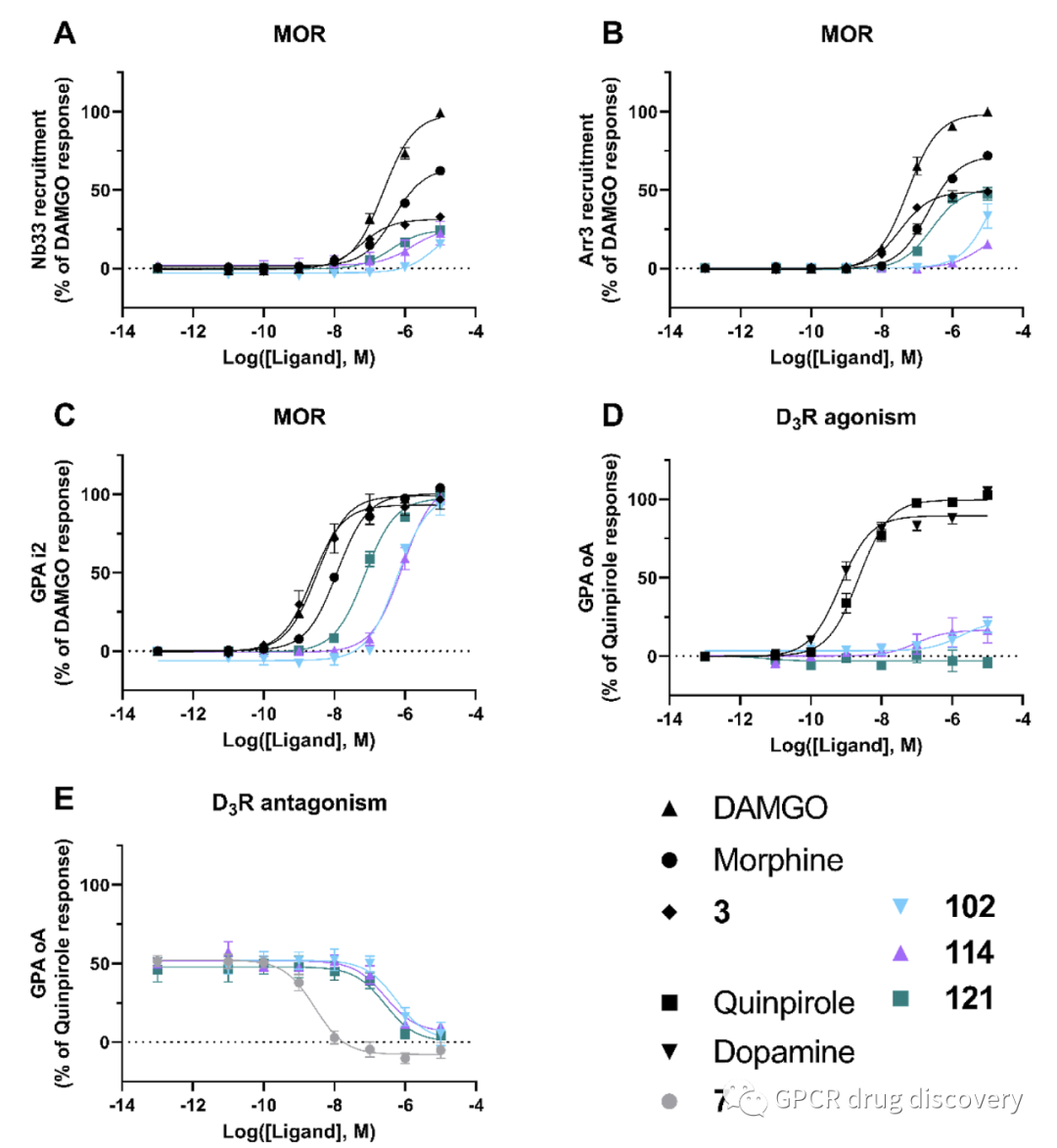
该系列的分子中,102、114、121均表现出完全激动 μ-阿片受体的活性 (Gi),部分激动Arr3活性;102和114 具有较弱的 D3R 激动活性,其中121 具有较强的 D3R 拮抗剂性质。
最后,作者用分子对接的方法,对多个分子与μ-阿片受体和 D3R 的激动活性进行了分析。
结论:
该研究中,作者将满足多受体亲和性、亚型选择性和探索创新化学空间的药物设计特点结合起来,从而在两个受体靶点上都保留药理学上所期望的疗效,同时还尝试优化外周与中枢神经系统活性,是一项具有挑战性的工作。
提高中枢神经系统穿透性的化学修饰往往不利于双重受体识别,而高度精细的母核可以利用基于结构的药物设计的细微差别,将药物设计推向极高的亲和力和效力极限,但往往不利于脑/血浆分布和中枢神经系统活性。归根结底,关键在于能否找到适当的平衡。
这些新线索为体内镇痛提供了潜力,同时降低了成瘾性,可能为开发更安全的镇痛药物提供了新方向。
参考文献:
1. Bonifazi, A., Saab, E., Sanchez, J., Nazarova, A.L., Zaidi, S.A., Jahan, K., Katritch, V., Canals, M., Lane, J.R., and Newman, A.H. (2023). Pharmacological and Physicochemical Properties Optimization for Dual-Target Dopamine D3 (D3R) and μ‑Opioid (MOR) Receptor Ligands as Potentially Safer Analgesics. J. Med. Chem. 10.1021/acs.jmedchem.3c00417
Copyright© 上海博璞诺科技发展有限公司 2016-2019 沪ICP备16015171号 中国化工网 全球化工网 生意宝 著作权声明
地址:上海市浦东新区张江高科技园区海科路100号9B楼四楼 电话:+86-21-20608178 传真:+86-21-20608171 联系我们

