Detail
Source: https://pubmed.ncbi.nlm.nih.gov/33510026/
Abstract
Vicinal dibromides and dichlorides are important commodity chemicals and indispensable synthetic intermediates in modern chemistry that are traditionally synthesized using hazardous elemental chlorine and bromine. Meanwhile, the environmental persistence of halogenated pollutants necessitates improved approaches to accelerate their remediation. Here, we introduce an electrochemically assisted shuttle (e-shuttle) paradigm for the facile and scalable interconversion of alkenes and vicinal dihalides, a class of reactions that can be used both to synthesize useful dihalogenated molecules from simple alkenes and to recycle waste material through retro-dihalogenation. The reaction is demonstrated using 1,2-dibromoethane, as well as 1,1,1,2-tetrachloroethane or 1,2-dichloroethane, to dibrominate or dichlorinate, respectively, a wide range of alkenes in a simple setup with inexpensive graphite electrodes. Conversely, the hexachlorinated persistent pollutant lindane could be fully dechlorinated to benzene in soil samples using simple alkene acceptors.
Vicinal dibromides and dichlorides have found widespread applications as flame retardants, pest control agents, polymers, and pharmaceuticals (1, 2). They also serve as versatile synthetic intermediates in organic chemistry because of the inherent reactivity of carbon–halogen bonds (3, 4). Despite these attractive features, the preparation of dihalogenated molecules still mainly relies on the use of highly reactive and corrosive halogenating reagents such as Cl2 and Br2, which are hazardous compounds to transport, store, and handle (4–7). Two general strategies have been commonly used to avoid the direct use of Cl2 and Br2. The first strategy makes use of carrier reagents such as Et4NCl3 or pyridinium tribromide as bench-stable surrogates. Despite their increasing stability, they still require the use of X2 reagents for their syntheses and tend to be unstable and corrosive themselves because they are designed to readily release the corresponding X2 reagents (5–7). The second strategy relies on the in situ generation of the active halogenating species from the reaction between halides and strong oxidants, a feature that can limit the functional group (e.g., alcohols) compatibility of these reactions (5–7). The use of strong oxidants also creates thermodynamic challenges for the development of the reverse reactions, retro-dihalogenations, that could be applied to remediation of persistent halogenated pollutants.
Transfer hydrofunctionalization proceeding through a shuttle catalysis (8) paradigm has emerged as a powerful and versatile strategy to reversibly functionalize and defunctionalize organic molecules without using or releasing hazardous reagents (8–15) such as HCN (9). However, catalytic and reversible transfer reactions have so far been limited to alkene monofunctionalization (16) reactions, which usually involve the transfer of an HX molecule (8, 15). By contrast, the synthetically appealing simultaneous transfer of two functional groups in a catalytic reversible transfer difunctionalization process has remained largely elusive despite the vast synthetic potential of these reactions in organic synthesis. In particular, reactions involving the formal transfer of extremely reactive and corrosive molecules such as Cl2 (17, 18) and Br2 from easier-to-handle, stable bulk chemicals such as inexpensive 1,2-dichloroethane and 1,2-dibromoethane would be highly desirable because of the widespread synthetic applications of dihalogenated molecules in flame retardants, pesticides, materials, and natural products (1, 2, 19) (Fig. 1A). The inherent reversibility of such a shuttle reaction would further unlock the facile retro-dihalogenations of end-of-life halogenated products for remediation (Fig. 1A).
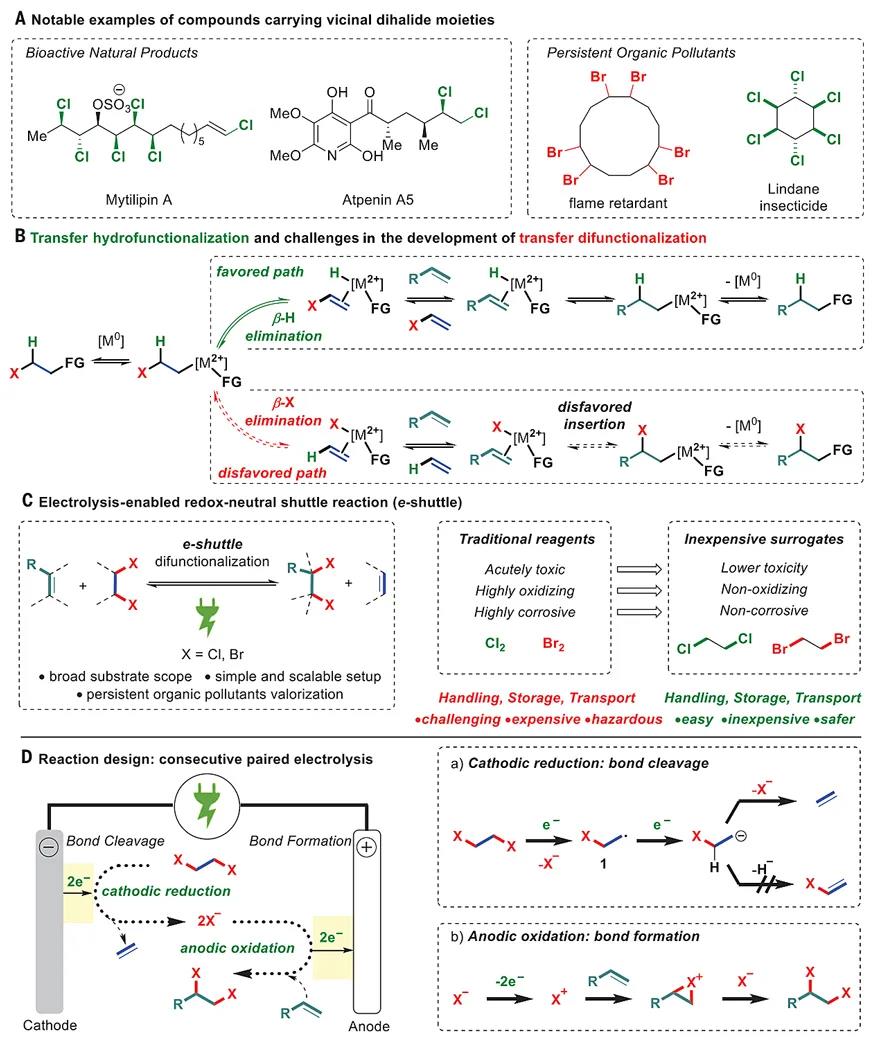
Fig. 1 Reaction design and challenges of transfer difunctionalization.
(A) Notable examples of compounds carrying vicinal dihalide moieties. (B) Transfer hydrofunctionalization and challenges in the development of transfer difunctionalization. (C) Electrolysis-enabled redox-neutral shuttle reaction (e-shuttle). (D) Reaction design showing consecutive paired electrolysis.
The challenge in developing transfer difunctionalizations such as transfer dihalogenations originates from the catalytic approach generally used in shuttle catalysis. Transfer hydrofunctionalizations such as hydrocyanation (9) rely on the intermediacy of an alkyl-metal complex that readily undergoes fast and reversible β-hydride elimination, thus triggering the transfer of a hydrogen atom alongside the desired functional group (15) (Fig. 1B). Unfortunately, the ease of β-hydride elimination makes the selective, competitive elimination of other synthetically useful groups challenging (20). Furthermore, while β-hydride elimination is a fast and reversible process, the subsequent migratory reinsertion of an alkene into a metal-halogen bond is often kinetically and thermodynamically disfavored because of the high stability of these types of bonds (6). Thus, a mechanistically distinct approach to favor halogen transfer over hydrogen transfer is crucial to unlock this important class of transfer difunctionalization reactions.
Electrosynthesis has recently experienced a renaissance in organic chemistry because it takes advantage of readily available electrical current as a sustainable and inherently safe redox reagent (21–24). Notable advances have been made in halogenation reactions (25, 26), as illustrated by an elegant example of dichlorination from Lin et al. (25). However, this reaction, as well as many other electrochemical reactions, has to be coupled to another sacrificial half-reaction, e.g., proton reduction to form hydrogen, at the counterelectrode (23, 24). In addition to this limitation, current protocols can often be further limited by the use of complex reaction setups including expensive metal electrodes (23, 24).
We envisaged that consecutive paired electrolysis (27) involving a domino reduction-oxidation cascade (28, 29), a class of ideal yet comparatively rare electrochemical reactions in which both electrodes are used in the desired transformation, could provide a path to reversible, electrochemically mediated shuttle reactions (e-shuttle). We surmised that the reversible cleavage of two strong carbon-halogen bonds through a controlled electron-transfer process initiated by a simultaneous, simple reduction and oxidation of key intermediates at the cathode and anode, respectively, would unlock this transformation (Fig. 1, C and D). In our hypothesis, the single-electron reduction of the dihalide at the cathode releases the X– anion and generates the carbon radical 1, which is almost instantly reduced again to generate a carbanion (30). As a central design, the subsequent selective loss of the second X– instead of a hydride breaks the C−X bond, releasing the alkene compound. Considering that a halide anion is a much better leaving group than a hydride, the competing undesired β–H elimination, which is often the preferred pathway when alkyl-transition metal complexes are involved as intermediates, can be effectively suppressed by this electrochemical approach. The subsequent oxidation of X– at the anode followed by reaction with the alkene delivers the desired product, which closes the cycle by reestablishing the C−X bonds in a fully isodesmic process. Precise control of the potential applied on the electrodes and the highly tunable cell voltage would make this strategy modular and versatile with regard to the group transferred. This is an advantage over the organometallic strategy, in which each shuttle reaction relies on a completely different combination of metal and catalyst requiring independent optimization campaigns (15).
At the outset of our investigations, a transfer dibromination was optimized in an undivided cell using inexpensive isostatic graphite as the electrode material under constant current conditions at room temperature, a reaction setup easily accessible to nonspecialized laboratories. 1,2-Dibromoethane (DBE) was selected as a formal Br2 donor because it is an inexpensive and stable reagent, produced on a bulk scale, that would release gaseous ethylene as a by-product, thus providing a driving force for the shuttle process. Most commercial suppliers offer this reagent at an even lower price (per mole of Br2 equivalents) than Br2 itself, presumably reflecting the challenges and costs inherent to transporting and storing the volatile and corrosive Br2 (31). Optimal results were obtained with 5 equivalents of 1,2-dibromoethane as the Br2 donor, 1 volume percent (vol %) 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) as a key additive (32), and 2 equivalents of Et4NBF4 as electrolyte in acetonitrile, providing the targeted 1,2-dibromide 2 in 84% yield (measured by integration of nuclear magnetic resonance spectra) when 3 F of electricity with respect to 1-dodecene was applied (Fig. 2). As indicated by cyclic voltammetry (CV) studies, the HFIP facilitates the reduction of the DBE donor and suppresses the undesired and unproductive reductive oligomerization and/or polymerization of alkene acceptors at the cathode (see Fig. 2Aand figs. S31 and S32 for more details). The supporting electrolyte can be easily crystallized from the reaction mixture to be recycled.
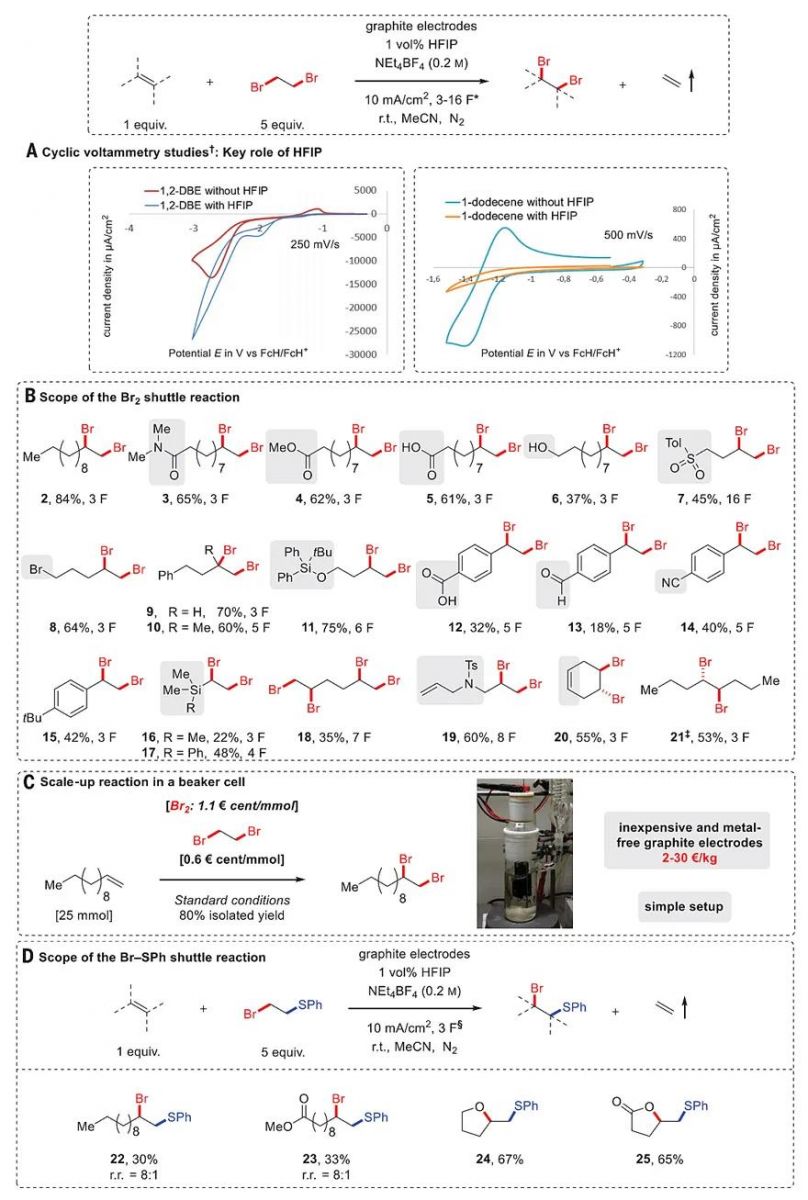
Fig. 2 Scope of the Br2 and Br−SPh shuttle reactions.
All yields are isolated yields of the products. (A) CV studies. (B) Scope of the Br2 shuttle reaction. (C) Scale-up reaction in a beaker cell. (D) Scope of the Br−SPh shuttle reaction. *1 F equals ~62 min of electrolysis time. †Conditions for CV studies: A 5 mM solution of 1,2-DBE and 1-dodecene in MeCN using NEt4BF4 (0.2 M) as the supporting electrolyte at a graphite electrode with and without 1 vol % HFIP as the additive. ‡(E)-4-octene used as the starting material. §1 F equals ~31 min of electrolysis time. r.t., room temperature; FcH, ferrocene; r.r., regioisomeric ratio.
Using this protocol, a broad range of unactivated terminal alkenes (2−11) were readily converted to the corresponding dibromide product in modest to good yields. The reaction conditions were compatible with a large variety of functional groups such as amide (3), ester (4), free carboxylic acid (5), primary alcohol (6), sulfone (7), and bromide (8). Only a small amount of the alcohol group oxidation to aldehyde was observed, confirming the mild nature of the reaction conditions. Activated alkenes, such as styrene (12–15) and vinyl silane (16 and 17), proved to be suitable substrates as well, albeit giving slightly lower yields because of undesired alkene oligomerization. Although hexa-1,5-diene underwent twofold 1,2-dibromination to yield the tetra-brominated product 18 in acceptable yield, selective mono-1,2-dibromination was observed for several other unconjugated dienes (19 and 20). (E)-4-Octene was smoothly dibrominated to produce the meso-dibromide 21 as the single diastereomer. To demonstrate the scalability and robustness of this e-shuttle process, the transfer bromination of 1-dodecene was readily scaled up to a 250-ml beaker cell from a 10-ml reaction vial to give 6.57 g (80% yield) of product 2 under otherwise identical reaction conditions (Fig. 2C).
Taking advantage of the reversible elimination of a −SR group (33), we could next also develop a transfer bromothiolation of alkenes to prepare 1,2-bromothioether derivatives, which are valuable synthetic intermediates usually accessed through multistep synthesis involving highly reactive RSBr reagents (34, 35). Several terminal alkenes were successfully converted to the targeted bromothioether product under otherwise identical conditions, taking 2-bromoethyl phenyl sulfide (5 equivalents) as the PhS−Br donor (Fig. 2D). The lower yields arose from competing formation of vicinal disulfides and RS−SR, as well as alkene oligomerization. The ester (23) functional group was compatible. An interrupted shuttle reaction took place when pent-4-en-1-ol and pent-4-enoic acid were used as the substrates, delivering the cyclic ether (24) or lactone derivatives (25) through subsequent intramolecular nucleophilic attack, demonstrating the method’s potential for the development of cascade reactions.
We next explored a transfer dichlorination reaction (Fig. 3). 1,2-Dichloroethane (DCE) was selected as the donor because it is an inexpensive bulk chemical (20 million tons/year) that is produced as a central intermediate in polyvinylchloride (PVC) production using the excess of Cl2 gas generated during the Chlor-alkali electrolysis process (36). The desired dichloride 28 was obtained in 40% yield when 5 mol % of manganese(II) chloride tetrahydrate was introduced as a mediator (25) using an otherwise identical electrochemical setup to the dibromination protocol. The yield was further increased to 70% when DCE (~125 equivalents) was used as the solvent (37). Although this procedure was efficient for a wide set of terminal alkenes (28–34; Fig. 3B), it failed for more challenging 1,1,2-trisubstituted alkene 26 (Fig. 3A), a feature largely attributable to the undesired 1,2-dechlorinative decomposition of the product 27 and alkene oligomerization of the starting material through cathodic reduction. We reasoned that these two challenges could be smoothly addressed by choosing a more suitable dichloride donor. On the basis of the known reduction potentials of a large set of simple chlorinated compounds (38), we hypothesized that polychlorinated C2 donors, which are more readily reduced, should lead to a more favorable reaction outcome. Experimentally, an excellent correlation between the reduction potential of a series of donors was indeed observed, leading to the identification of 1,1,1,2-tetrachloroethane, a stable, noncorrosive compound, as the reagent of choice, affording the desired dichloride product 27 in 90% nuclear magnetic resonance (NMR) yield (Fig. 3A). Using this procedure, a series of monosubstituted, disubstituted, and trisubstituted alkenes participated smoothly in the 1,2-transfer dichlorination reaction, with free alcohol (29, 35, and 37), ester (30), imide (32), phosphonate (33), sulfone (34), and Ts- and Boc-protected amine moieties (38 and 39) proving compatible. An internal alkyne (37) was even partially compatible with the reaction conditions despite the minor formation of unidentified by-products. Various styrene-derived alkenes were converted to the corresponding 1,2-dichlorides in good to excellent yield (42–52), leaving the Br, Cl, CN, CF3, CHO, and COOH functional groups untouched. Indene was diastereoselectively transformed into trans-1,2-dichloride 50 (diastereomeric ratio > 19:1). Both (E)- and (Z)-1-phenylpropene were converted to the anti-dichloride 51 in similarly high diastereoselectivity. The 1,2-dichloride compound 52, bearing a reactive benzylic tertiary C−Cl bond, was prepared in good yield from α-methylstyrene. Several other activated alkenes, such as the silyl- and ester-substituted alkenes, also proved to be viable substrates to deliver the dichloride products (53–56), in particular, methyl cinnamate was converted to the 1,2-dichloride 56 in an excellent diastereomeric ratio (>19:1). To our delight, preliminary experiments showed that this protocol can be readily extended to the 1,2-chlorothiolation transfer reaction using the commercially available 2-chloroethyl phenyl sulfide (10 equivalents) as the donor (Fig. 3C). The lower yield can be explained by the undesired formation of RS−SR species as well as substrate oligomerization.
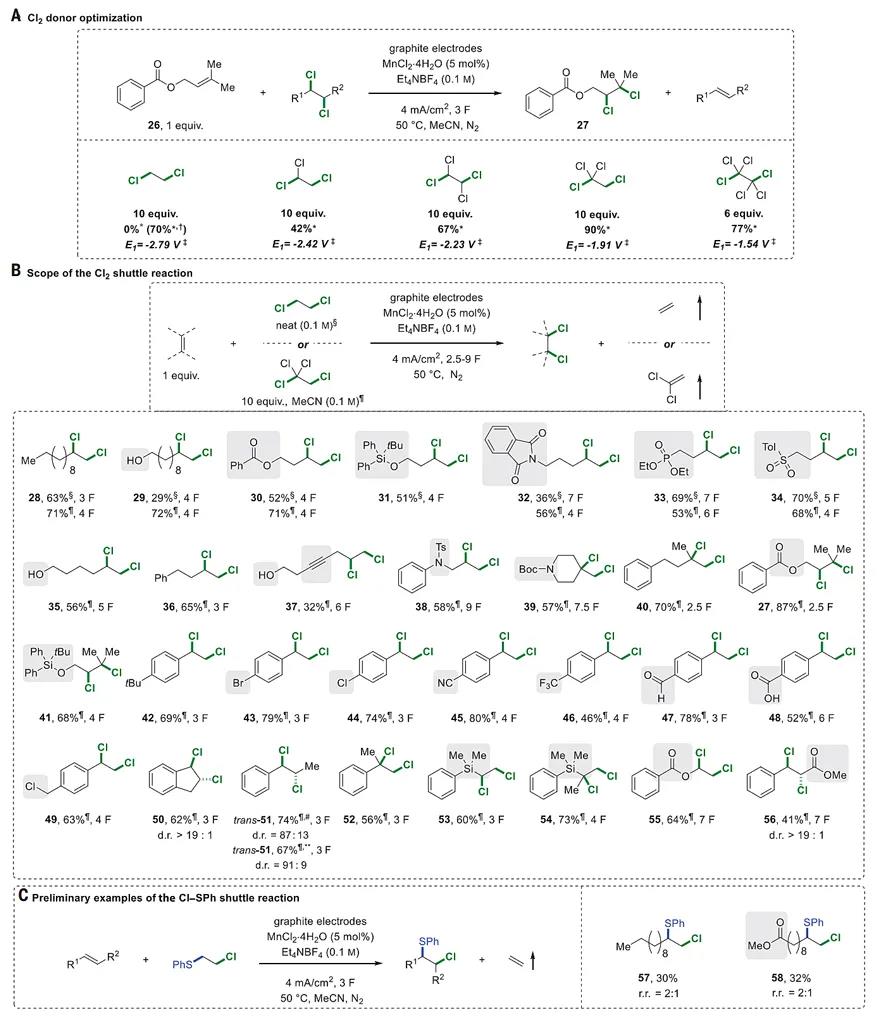
Fig. 3 Scope of the Cl2 and Cl−SPh shuttle reactions.
Here, 1 F equals ~81 min of electrolysis time. All yields are isolated yields of the products unless otherwise noted. (A) Cl2 donor optimization. (B) Scope of the Cl2 shuttle reaction. (C) Preliminary examples of the Cl−SPh shuttle reaction. *1H-NMR yield with mesitylene as the internal standard. †Neat DCE (0.1 M) as the donor and 1-dodecene (1 equivalent) as the acceptor. ‡Redox potential (V versus SCE) measured for the first reduction peak at 0.2 Vs−1 for polychlororethanes (2 mM) in DMF + 0.1 M (C3H7)4NBF4 at a glassy carbon electrode according to (38). §Neat DCE (0.1 M) as the donor. ¶Reaction performed in MeCN (0.1 M) with 1,1,1,2-tetrachloroethane as the donor. #(E)-prop-1-en-1-ylbenzene used as the starting material. **(Z)-prop-1-en-1-ylbenzene used as the starting material. d.r., diastereomeric ratio.
In contrast to traditional halogenation methods, the inherent reversibility of the e-shuttle strategy offers a platform to develop retro-dihalogenation reactions. Given the environmental persistence of several halogenated compounds produced at commodity scale, such as flame retardants and insecticides, e-shuttle could facilitate their recycling and valorization through retro-dihalogenations, which could ultimately lead to a circular economy for these important chemicals. A notable example is lindane [gamma-hexachlorocyclohexane (HCH)], a compound that was once used worldwide as an effective broad-spectrum insecticide in crop protection, which is now classified as a persistent organic pollutant because of its high toxicity and high persistency in the environment (39–42). Global quantities of HCH wastes still present in the environment range between 4 and 7 million tons worldwide, highlighting the pressing challenge of finding methods to recycle and remove this compound from contaminated soils (39). We thus questioned whether this waste material, which, among other chemical and biological approaches (40), can only be inefficiently degraded through normal electrochemical recycling methods (43–45), could be efficiently retro-dihalogenated using our e-shuttle strategy (Fig. 4A). Indeed, lindane, through three successive retro-dichlorination events, successfully transferred its six chlorine atoms to an acceptor alkene to form benzene, the fully dechlorinated by-product of lindane, alongside a dichlorinated alkane. Five illustrative alkene examples gave excellent yields up to 89% (>95% gas chromatography yield of benzene; Fig. 4B), demonstrating the generality of this process and the possibility of accessing a wide variety of potentially useful chemicals. A scale-up experiment (87.5 mmol of alkene) further showcased the efficiency of the retro-dichlorination process (Fig. 4C). The exceptional functional group tolerance of our e-shuttle strategy made us next question whether we could successfully retro-dichlorinate lindane–contaminated soils through a transfer dichlorination reaction (Fig. 4D). In theory, such a process could lead to a new avenue to chemically remediate highly contaminated soils, which are mainly caused by leachates of improper disposal at landfilling or dump sites (39, 40, 42), through simultaneous synthesis of commercially relevant dichlorinated chemicals. This could provide an alternative to current industrial approaches that focus on the generation of HCl from chlorinated waste (42).
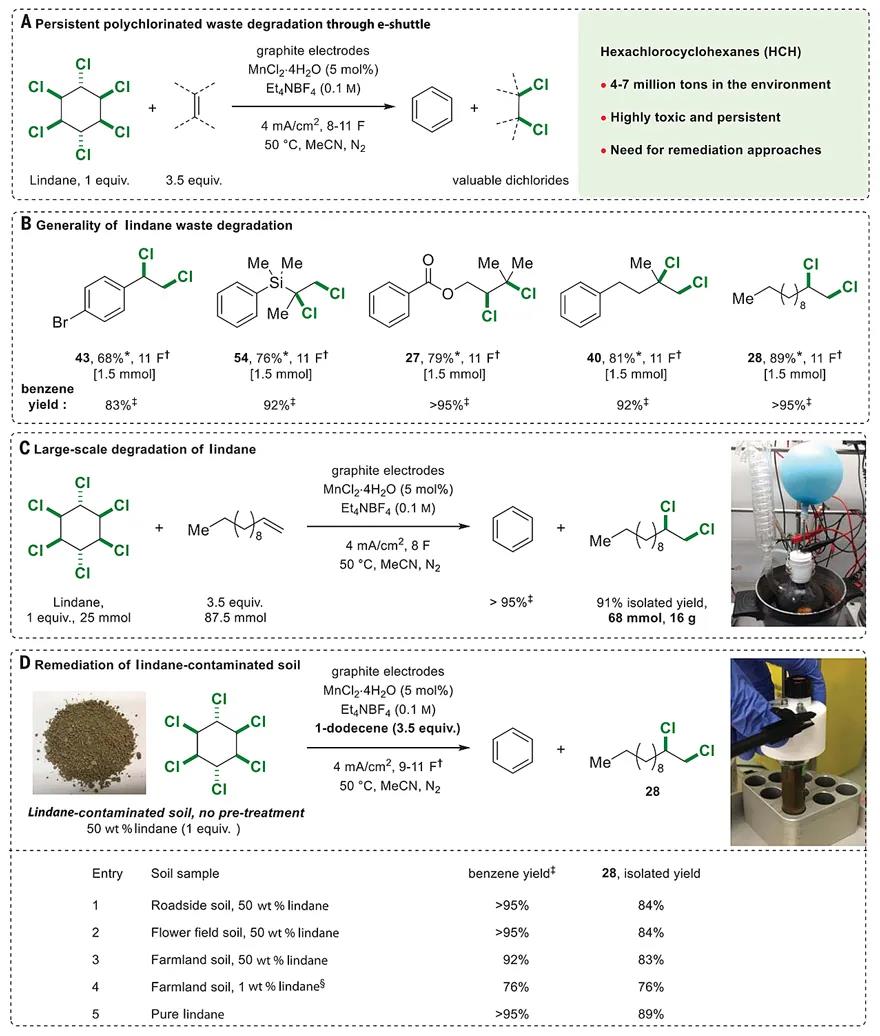
Fig. 4 Application of e-shuttle reactions.
(A) Persistent polychlorinated waste degradation through e-shuttle. (B) Generality of lindane waste degradation. (C) Large-scale degradation of lindane. (D) Remediation of lindane-contaminated soil. *Isolated yield. †1 F equals ~81 min of electrolysis time. ‡Yield measured by gas chromatography using mesitylene or anisole as the internal standard. §Lindane was extracted with MeCN before degradation.
Artificially contaminated soils are commonly used as models in environmental chemistry to perform proof-of-concept chemical remediation experiments (46). Therefore, to mimic the composition of soils contaminated by high concentration of HCH, three soil samples from different locations near our university campus, i.e., roadside, flower field, and farmland, were collected and homogeneously mixed with commercially available lindane. The 50 weight % (wt %) lindane–contaminated soil could be used directly in the reaction without any preextraction or filtration, delivering both the benzene and dichloride product in excellent yields and high purity (Entries 1 to 3; Fig. 4D), a result comparable to the experiments using pure lindane (Entry 5; Fig. 4D). This result shows that our degradation process is compatible with the biological and mineral impurities present in three different soil types. Although the exact composition of environmentally relevant contaminated soils might differ from our samples (39, 41, 42), we nevertheless believe that these positive preliminary results using a variety of soil samples support the feasibility of this approach. A much lower lindane-soil ratio of 1 wt % was extracted with the reaction solvent before the degradation to also afford good yields for both benzene (76%) and dichloride (76%, Entry 4; Fig. 4D). This alternative preextraction protocol acts as a further proof-of-concept that might help in the design of larger-scale remediation processes in which undesired soil contamination with electrolytes and Mn catalysts can be prevented. The large-scale feasibility of an extraction approach has been demonstrated by the successful treatment of ~70,000 tons of HCH-contaminated soils in the Netherlands in a full-scale soil-washing plant, which achieved HCH removal efficiency of >99.7% (42).
Collectively, these preliminary results serve as a proof-of-principle for the direct remediation of lindane-contaminated soils using e-shuttle methodology.
Supplementary Materials
science.sciencemag.org/content/371/6528/507/suppl/DC1
Materials and Methods
Figs. S1 to S41
Tables S1 to S16
NMR Spectra
https://www.sciencemag.org/about/science-licenses-journal-article-reuse
This is an article distributed under the terms of the Science Journals Default License.
Copyright© VastPro (Zhejiang) Pharmaceutical Co.,Ltd. ChinaChemNet Toocle Copyright Notice
4th Floor, Building 9B, 100 Haike Road, Pudong New Area District, Shanghai, 201210 P. R, China.
Tel: +86-21-20608178 Fax: +86-21-20608171 Contact us


