Detail
Source:https://www.nature.com/articles/s41557-021-00640-2
Abstract
Electrochemical techniques have long been heralded for their innate sustainability as efficient methods to achieve redox reactions. Carbonyl desaturation, as a fundamental organic oxidation, is an oft-employed transformation to unlock adjacent reactivity through the formal removal of two hydrogen atoms. To date, the most reliable methods to achieve this seemingly trivial reaction rely on transition metals (Pd or Cu) or stoichiometric reagents based on I, Br, Se or S. Here we report an operationally simple pathway to access such structures from enol silanes and phosphates using electrons as the primary reagent. This electrochemically driven desaturation exhibits a broad scope across an array of carbonyl derivatives, is easily scalable (1–100 g) and can be predictably implemented into synthetic pathways using experimentally or computationally derived NMR shifts. Systematic comparisons to state-of-the-art techniques reveal that this method can uniquely desaturate a wide array of carbonyl groups. Mechanistic interrogation suggests a radical-based reaction pathway.
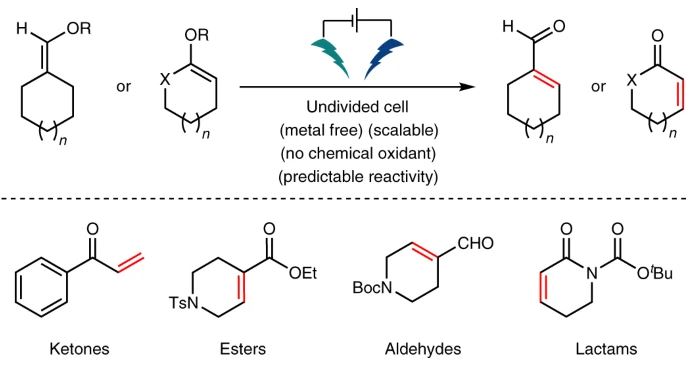
Main
The removal of one molecule of hydrogen adjacent to a carbonyl compound is one of the simplest organic oxidation reactions known and is a widely employed tactic in synthesis1,2,3. Classic methods to accomplish this transformation involve indirect α-functionalization approaches that traverse through halide, sulfur and selenium derivatives4,5,6,7,8. Chemoselective methods that directly afford enones from ketones are, indeed, more desirable and have been extensively explored (Fig. 1). Among them, the Saegusa–Ito reaction, discovered in 1978, remains the most oft-applied method for such applications9. In its canonical implementation, the formation of a silyl enol ether, followed by exposure to stoichiometric quantities (from 0.5 to 1.0 equiv.) of palladium delivers the desired α,β-desaturated product9. Variants that employ a co-oxidant (for example, O2, quinone or [Cu]) to lower the Pd levels are also reported10,11. Another popular approach involves the use of stoichiometric o-iodoxybenzoic acid (IBX) through a process based on single electron transfer12,13. Recently, two new methods have also appeared from the Newhouse and Dong groups that allow the use of catalytic amounts of palladium and copper, respectively14,15,16,17,18,19,20. These methods expand the scope of available desaturation methods to nitriles, esters, lactones and lactams and do not require the preparation of enol ethers. As the essence of this reaction involves a formal two-electron oxidation, it reasonable that even simpler redox approaches might be developed. Indeed, in 1973, the Shono group demonstrated that enol acetates can undergo anodic oxidation in AcOH as the solvent to afford the corresponding enone. To deliver synthetically useful yields of product, α-substitution was required with simple cyclohexanone substrates to provide <10% enone21,22. Similar reactivity was also observed by Moeller and co-workers in their studies of silyl enol ether alkylation where trace amounts of enone were isolated as a by-product (<5% yield)23,24,25. Building on these encouraging studies, we report herein an electrochemically driven approach to elicit desaturation that requires no metals or chemical oxidants and features a broad substrate scope with inherent scalability. The utility of this method is placed in the context of the most popular and recently disclosed methods, and a simple method for predicting reactivity is also described.
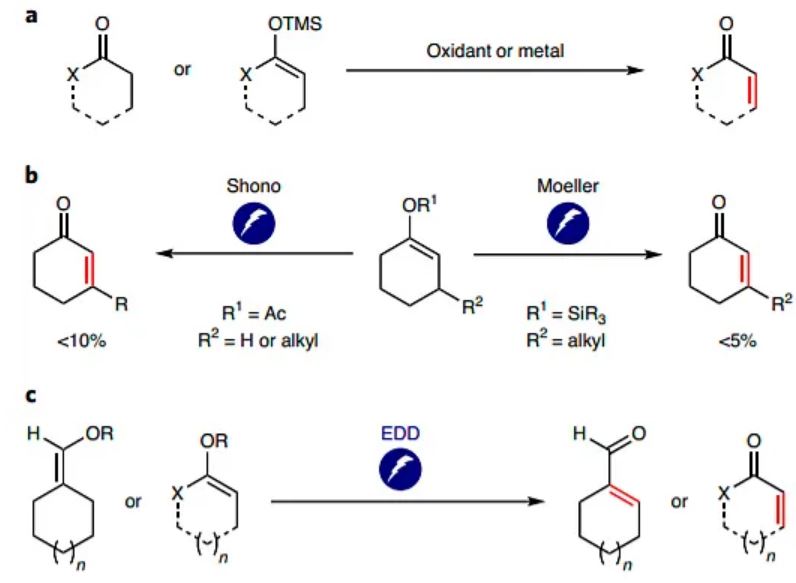
a, Chemical approaches: the most used chemical approaches for the desaturation of carbonyl and enol ether compounds. Ito et al. reported a method using stoichiometric palladium, which is limited to ketones and aldehydes9. Newhouse also reported studies using palladium, which also require expensive Zn(TMP)2 (bis(2,2,6,6-tetramethylpiperidinyl)zinc), and an elevated temperature14,15,16. Studies reported by Chen and Dong require copper, peroxide, and elevated temperature and have limited scope18. Previous studies from the Nicolaou lab use stoichiometric IBX, a strong oxidant and are limited to ketones and aldehydes12. b, Electrochemical precedents: the Shono group demonstrated that enol acetates can undergo anodic oxidation to afford the corresponding enone21,22. Moeller and co-workers observed that silyl enol ether can similarly undergo direct anodic oxidation to form trace amounts of enone under the described conditions23,24. In both cases, the methods are limited to ketones, have shown limited functional-group tolerance and low yields were obtained with non-substituted ketones. c, EDD: the metal, chemical oxidant free and scalable electrochemical desaturation method is described. EDD is applicable with various types of carbonyls—ketones, esters, lactams and aldehydes.
Results and discussion
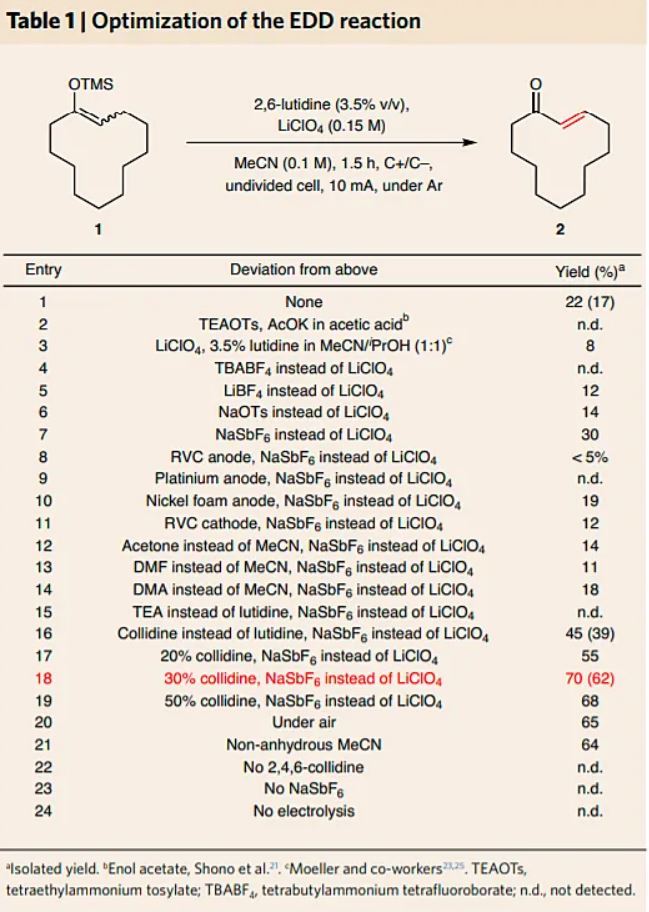
The tetramethylsilyl (TMS) enol ether of cyclododecanone 1 was chosen for the initial optimization of the electrochemically driven desaturation (EDD); an abbreviated summary is depicted in Table 1 (and see Supplementary Tables 1–5). Trial runs using the literature conditions noted above provided only trace quantities of the product. Our prior experiences of electrochemical reaction development served as a template for this study26,27,28,29,30,31,32,33. A myriad of electrolytes, electrodes and solvents were evaluated. First, an electrolyte screen revealed that inorganic non-nucleophilic salts proved optimal (Table 1, entries 4–7) with NaSbF6 (US$0.56 g–1) as they delivered the highest conversion. The use of a graphite anode was found to be essential, whereas several materials were suitable for the cathode (Table 1, entries 8–11). Ultimately, the low cost (~US$0.1 cm–2) and efficiency of graphite motivated its selection for both electrode materials. Of all the solvents screened, MeCN, acetone, dimethylacetamide (DMA) and dimethylformamide (DMF) could be employed, but MeCN gave the highest yield across a broad range of substrates. A variety of bases were also tested, and heteroaromatic amines proved most promising (Table 1, entries 15 and 16). 2,4,6-collidine (30% v/v, entry 18) emerged as the optimum as it provided the desired product 2 in a 62% isolated yield. The final set of EDD conditions tolerated exogenous air and moisture, led to completion in 90 minutes or less and can be set up in minutes using a simple undivided cell and a commercial potentiostat.
Table 1 Optimization of the EDD reaction
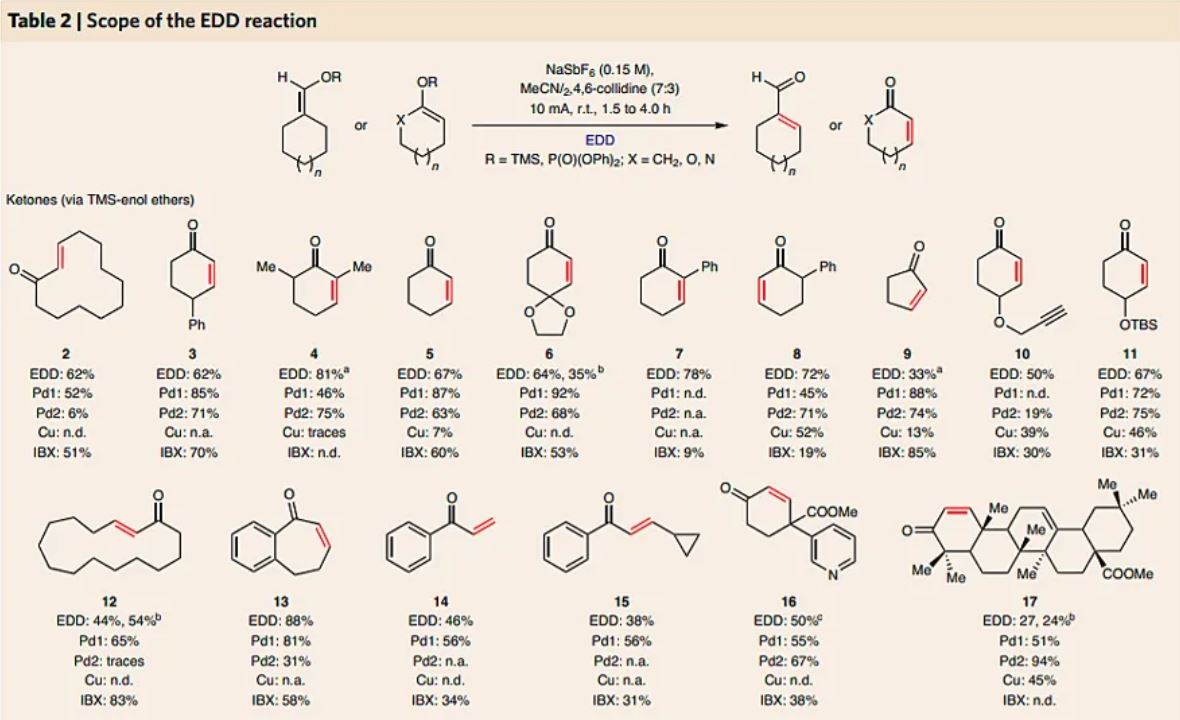
EDD could be applied to a diverse set of carbonyl derivatives, as illustrated in Table 2. As numerous desaturation methods are available to the practitioner, all of the results are placed in context by direct comparison to the powerful Pd, Cu and hypervalent iodine-based systems. With regards to ketone substrates, both cyclic (from 5- to 15-membered rings) and acyclic derivatives could be employed (2–17). This stands in contrast to recently developed catalytic methods that operate smoothly on cyclic systems but fail on acyclic ones (14 and 15)15,18. As the EDD of ketones is reliant on the formation of a silyl enol ether, regioselective desaturation is possible simply by tuning the conditions (that is, 7 versus 8). Substituents at the α-, β- and γ-positions are all tolerated, as well as Lewis-basic heteroatoms (16), alkynes (10), proximal cyclopropanes (15), esters (16 and 17), tert-butyl(dimethyl)silyl (TBS)-protected alcohols (11) and acid-labile ketals (6). A two-step in situ EDD protocol was also developed to afford enones 6, 12 and 17 in decent yields directly from the respective ketone starting material. Esters and lactones, substrate classes that have only recently succumbed to direct dehydrogenation14,17,18, can also be subjected to EDD using the corresponding diphenylphosphate ester derivatives (18–31). Such enol derivatives are easily prepared and hydrolytically stable, unlike the corresponding silyl ketene acetals. Simple lactones and benzolactones, which are outside the substrate scope of the IBX and Saegusa methodologies, can be smoothly dehydrogenated. As with the EDD of ketones, the functional-group tolerance here is also broad and includes aryl halides (22 and 24), CF3 (30), oxidizable anisoles (23), tosyl-protected amines (29) and alkenes (32). In addition, α-aryl lactones also afforded the desired products in satisfactory yields (21–23 and 25). The difficulty of desaturating such substrates has been documented by Dong18. They are often alternatively accessed through cross-coupling on the corresponding vinyl halide derivatives or through α-bromination/elimination sequences34,35. Note that in comparing EDD with other precedented methods, the set of conditions reported by the Newhouse group was found to provide the desaturated α-aryl lactones in moderate yields. Next, the particularly difficult class of aldehydes were investigated (32–34), with silyl enol ethers employed). Owing to the instability of such desaturated products, the yields observed were moderate (and accompanied by 5–11% of recovered parent aldehyde). Other direct catalytic methods for this dehydrogenation are not applicable, with the Saegusa protocol being the only other option. Lactams, a similarly challenging class of carbonyls, were surveyed as diphenylphosphate-ketenimine acetals, and in select cases (35–37) were viable.
Table 2 Scope of the EDD reaction
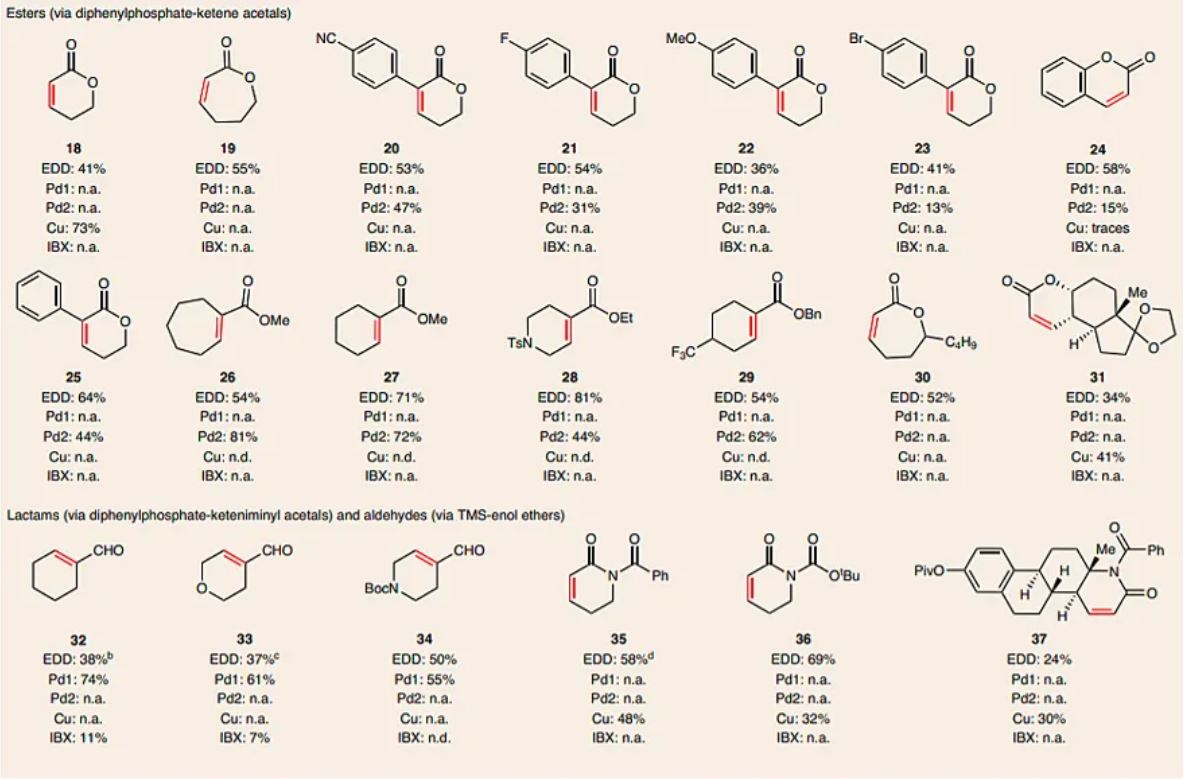
The scalability of the method was evaluated using cyclopentadecanone-derived silyl enol ether 38 on a 4 g scale (Fig. 2a) to afford enone 12, a key intermediate in the synthesis of (R)-muscone 39, a valuable ingredient in the fragrance industry36. A simple increase of current (from 10 to 300 mA) and the use of alternating polarity (to avoid any accumulation of material at the anode) enabled the standard EDD reaction to smoothly deliver compound 12 in a 66% yield. To increase the scale further, the design and assembly of a flow apparatus that contained six reaction cells was undertaken (Fig. 2a). After optimization, 100 g of 38 were successfully converted into compound 12 by increasing the current value to 3.6 A (compared to 300 mA in batch) to obtain a 61% isolated yield and 27% recovered starting material 38.
Fig. 2: Scale-up and mechanistic study of EDD reaction.
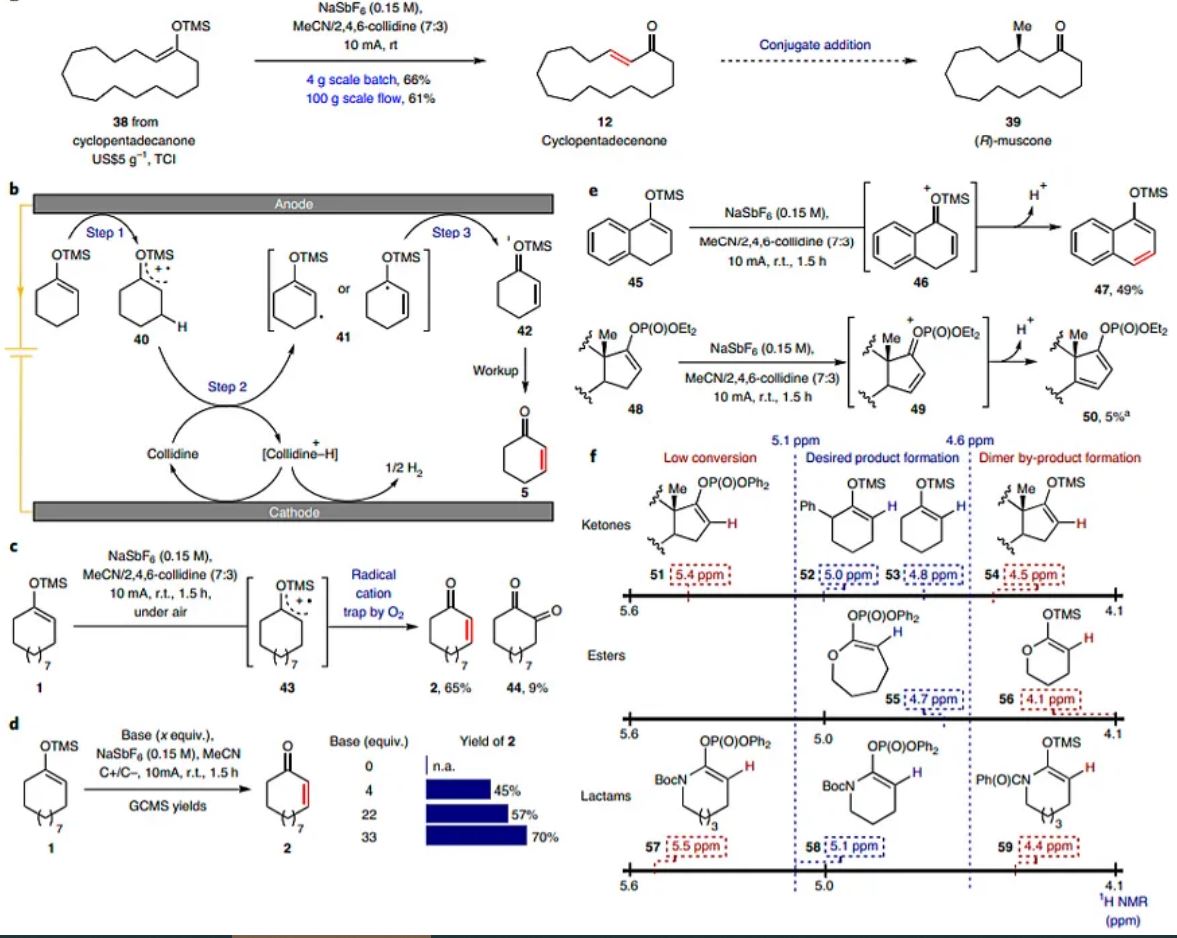
a, Batch (4 g) and flow (100 g) scale-up of a key intermediate in the synthesis of (R)-muscone36. TCI, Tokyo Chemical Industry. b, Proposed mechanism of the EDD reaction based on observations. It is postulated that EDD proceeds through three elementary steps: step 1, formation of the radical cation intermediate through direct anodic oxidation; step 2, deprotonation of the radical cation intermediate to afford the allylic radical and step 3, a second oxidation to form an oxonium intermediate. c, Experimental evidence for step 1. d, Experimental evidence for step 2. e, Experimental evidence for step 3. f, Prediction of reactivity based on 1H NMR chemical shift. aYield based on liquid chromatography mass spectrometry (LCMS) conversion. r.t., room temperature. GCMS, gas chromatography mass spectrometry conversion.
From a mechanistic standpoint, the EDD reaction accomplishes the formal removal of two electrons and one proton from the corresponding silyl enol ether. The electrochemical oxidation of silyl enol ethers was previously disclosed by Moeller23,24 and studied mechanistically by Wright and co-workers25. These studies demonstrated that the initial anodic oxidation leads to the formation of an enol ether radical cation intermediate by using a cyclopropyl ring-opening clock. A similar conclusion was made by Moeller and co-workers when they oxidized various alkyl-enol ethers and thio-enol ethers37. It is therefore postulated that EDD proceeds through three elementary steps: (1) the formation of the radical cation intermediate 40, (2) deprotonation to afford 41 and (3) a second oxidation to form oxonium 42, which affords the desired enone product 5 (Fig. 2b). To provide empirical support for the proposed mechanism, three control experiments were designed and tested (Fig. 2c–e). First, the standard reaction conditions under air revealed the formation of the 1,2-diketone side product 44 in 9% yield. The amount of this by-product decreased to 4% when the reaction was performed under an inert atmosphere. In addition, the parent ketone was the only product observed when water was added to the reaction. These results suggest that the formation of compound 44 derived from molecular oxygen via a radical-type mechanism. Next, to support the crucial role of the base in the deprotonation event, the EDD reaction conditions were applied to compound 1 with various amounts of 2,4,6-collidine (Fig. 2d). No desired product was obtained when the base was excluded, which reinforces the importance of the base for the EDD reaction and its implication in the deprotonation step 2 (Fig. 2b). Furthermore, a noticeable improvement was observed between the reaction efficiency and base concentration. Finally, a third experiment was conducted to explore the formation of an oxonium intermediate (Fig. 2e). Compound 45 was subjected to the reaction conditions and afforded naphthalene 47 in a 49% yield. The formation of this product is in accordance with the formation of intermediate 46, which, after elimination, would deliver 47. Similarly, when the diethylphosphate 48 derived from dehydroepiandrosterone was subjected to EDD, diene 50 was also observed as a side-product.
During these studies it was empirically noted that the ketone, lactam and lactone substrates with vinylic proton shifts that registered between 4.6 and 5.1 ppm showed a good conversion to the desaturated product. However, compounds with NMR shifts lower than 4.6 ppm led to the formation of the dimer, hydrolysis and other by-products. Compounds with NMR shifts higher than 5.1 ppm showed a low reactivity toward oxidation. Based on these findings and Moeller’s reports37, it is possible to identify a trend between the oxidation potential of the enol ether (to form a radical cation intermediate similar to 40) and the electron density of the π-system (Fig. 2f and see Supplementary Fig. 10 for details). Compounds such as dehydroepiandrosterone-phosphate 51 and the eight-membered lactam 57 cannot be oxidized under the EDD reaction conditions due to the deshielded vinylic proton, which correlates to a higher oxidation potential (cyclic voltammetry (CV) = 2.06–2.47 V). When the vinylic proton was shielded, the oxidation potential became lower and reactivity towards EDD was observed (CV = 1.54–1.72 V). However, compounds such as dehydroepiandrosterone-TMS 54, lactone-TMS 56 and lactam-TMS 59, whose vinylic protons are also shielded, afforded low amounts of the desaturated product (CV = 1.32–1.4 V). In this case, the enol derivatives reacted as nucleophilic radicals rather than as electrophilic radical cations due to the greater cationic stabilization, which renders the EDD pathway less favourable38,39. Accordingly, this explains why electron-withdrawing enol-phosphates are required for esters and lactams in EDD rather than the corresponding silyl enol ethers.
Based on these observations, one can imagine predicting the outcome of the EDD reaction by calculating the NMR shift of the TMS enol ethers of interest (Fig. 3, step 1). With this idea in mind, a simple protocol using GAUSSIAN16, a quantum chemical calculation software, was developed. This protocol gave access to a calculated NMR shift based on the shielding constants using the gauge-including atomic orbital method in the WP04 database (functional) with an aug-cc-pVDZ basis set using tetramethylsilane as a reference (Fig. 3, step 2)40,41. Next, to obtain a more accurate value, the NMR shift value was corrected using an experimentally generated linear regression (Fig. 3, step 3 and see Supplementary section ‘Computational calculations using GAUSSIAN16’ for detailed graphical step-by-step guide). Finally, the corrected NMR shift can be used to predict the efficiency of the EDD reaction (Fig. 3, step 4). To our knowledge, this is a rare example of using a calculated NMR shift to predict the scope of an organic methodology42.
Fig. 3: Gaussian computational experiment to assess the feasibility of the EDD reaction; case study of TMS- enolates.
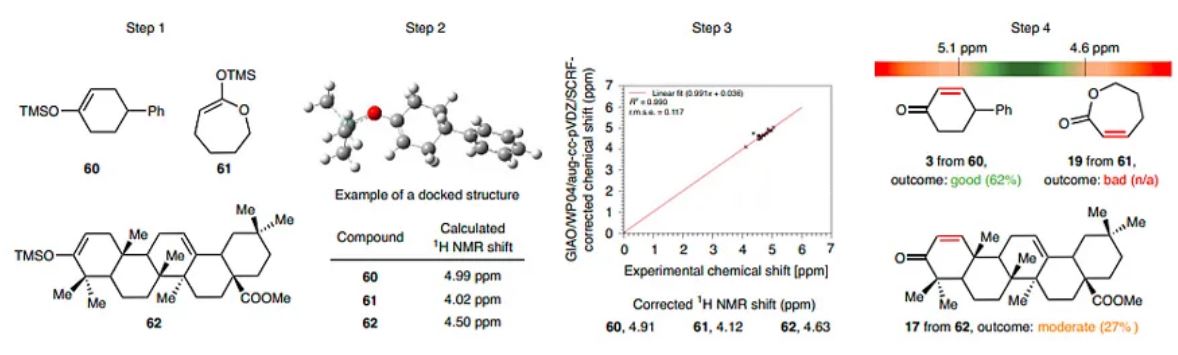
A simple four-step protocol using GAUSSIAN16 is described to predict the efficiency of the EDD reaction. Step 1, prepare a list of TMS enol ether substrates of interest. Step 2, use the Gaussian software GIAO/WP04/aug-cc-pVDZ//B3LYP/6-31 + G(d,p) functional to calculate the 1H NMR chemical shift of the desired enol ether. Step 3, use the experimentally generated linear regression to correct the calculated 1H NMR shift of the enol ether. Step 4, predict the outcome of the EDD reaction. r.m.s.e., root mean square error.
The desaturation of carbonyl derivatives is a basic reaction of utmost utility in organic chemistry, as it unlocks a variety of useful downstream transformations. Studies in this area continue to the present day; the contribution reported here affords a potentially simple solution to this problem. Drawing from early studies in electrosynthesis and more recent mechanistic studies of anodic enol-oxidation, a useful protocol for EDD was uncovered. This oxidation protocol can be performed in an undivided cell, on multiple scales, without the strict removal of air or water and in the absence of expensive metals, ligands or stoichiometric organic oxidants. As with the oxidation of alcohols, for which numerous methods are available, here this desaturation study is placed into context with the most powerful methods currently available to aid the practitioner. Finally, a simple 1H NMR-based rubric was created to allow users to experimentally or computationally predict which substrates are suitable for EDD, which should facilitate its rapid adoption.
Copyright© VastPro (Zhejiang) Pharmaceutical Co.,Ltd. ChinaChemNet Toocle Copyright Notice
4th Floor, Building 9B, 100 Haike Road, Pudong New Area District, Shanghai, 201210 P. R, China.
Tel: +86-21-20608178 Fax: +86-21-20608171 Contact us


