Detail
Abstract
Voltage-gated sodium (Nav) channels are targeted by a number of widely used and investigational drugs for the treatment of epilepsy, arrhythmia, pain, and other disorders. Despite recent advances in structural elucidation of Nav channels, the binding mode of most Nav-targeting drugs remains unknown. Here we report high-resolution cryo-EM structures of human Nav1.7 treated with drugs and lead compounds with representative chemical backbones at resolutions of 2.6-3.2 Å. A binding site beneath the intracellular gate (site BIG) accommodates carbamazepine, bupivacaine, and lacosamide. Unexpectedly, a second molecule of lacosamide plugs into the selectivity filter from the central cavity. Fenestrations are popular sites for various state-dependent drugs. We show that vinpocetine, a synthetic derivative of a vinca alkaloid, and hardwickiic acid, a natural product with antinociceptive effect, bind to the III-IV fenestration, while vixotrigine, an analgesic candidate, penetrates the IV-I fenestration of the pore domain. Our results permit building a 3D structural map for known drug-binding sites on Nav channels summarized from the present and previous structures.
Introduction
Voltage-gated sodium (Nav) channels govern membrane excitability in neurons and muscles1,2. Nine subtypes of Nav channels, Nav1.1-Nav1.9, are responsible for firing electrical signals with tissue specificity3. Consistent with their fundamental physiological significance, aberrant activities of Nav channels are linked to a wide range of pathogenic conditions. Nav channels are directly targeted by a number of FDA-approved anti-epilepsy drugs (AEDs), painkillers, and antiarrhythmic agents4,5,6,7,8.
High-resolution (cryo-EM) structures of eukaryotic Nav channels reported in recent years offered an advanced understanding of the mode of action (MOA) of some of the drugs at atomic level. For instance, the antiarrhythmic drugs, flecainide, quinidine, and propafenone are all accommodated in the central cavity of the pore domain (PD) of Nav1.5, but with non-overlapping binding poses9,10,11. Bulleyaconitine A (BLA), an active ingredient isolated from Aconitum plant for pain management and for the treatment of rheumatoid arthritis in China, cuts off the ion permeation by sitting right below the selectivity filter (SF) in the structure of Nav1.312. Nonetheless, the accurate binding mode of most of the Nav-targeting drugs is yet to be unveiled. Such information will not only reveal the chemical basis for the MOA of the clinically applied drugs, but also lay an important foundation for future drug development13.
We sought to identify the druggable sites on Nav channels by carrying out a systematic structural investigation using human Nav1.7-β1-β2 as the scaffold14,15. Nav1.7, encoded by SCN9A and primarily localized to the dorsal root ganglion neurons, has been explored as a prominent target for pain management16. Loss of function of Nav1.7 leads to indifference to pain17,18,19, whereas mutations that enhance channel activities are found in patients with primary erythromelalgia, paroxysmal extreme pain disorder and small fiber neuropathy20,21,22,23. Despite extensive efforts from a number of preeminent pharmaceutical companies, most of the Nav1.7-targeting drug candidates failed to meet the endpoint(s) of phase II trials8,24,25,26. We reckoned that structural details of Nav1.7 in complex with clinically applied drugs and lead compounds may afford a molecular insight that will benefit rational drug design or/and optimization.
In the structures of eukaryotic Nav channels, the core α subunit, which generally consists of 1500-2200 residues, is well resolved for the transmembrane (TM) and extracellular regions12,15,27,28,29,30,31,32,33,34. The four 6-TM (S1-S6) containing repeats exhibit a pseudo symmetry33,35,36. The S1-S4 segments in each repeat constitute the flanking voltage-sensing domain (VSD), and the S5-S6 segments enclose the PD that is responsible for selective and gated ion permeation28,33,34,37. Above the PD, several heavily glycosylated extracellular loops (ECL) are stabilized by multiple disulfide bonds32,38.
To explore potential druggable sites, we selected representative drugs and lead compounds with diverse chemical backbones (Supplementary Table 5). Our efforts were unfortunately limited by the availability of the chemicals. Here we present cryo-EM structural analysis of Nav1.7 treated with the following drugs: a local anesthetic drug bupivacaine (Marcaine) and a chemically related AED lacosamide (Vimpat), an anticonvulsant drug carbamazepine (Tegretol) that is also used for the treatment of trigeminal neuralgia and epilepsy39,40,41, a synthetic vincamine derivative vinpocetine (Cavinton) that can target Nav channels for the treatment of cerebrovascular disorders, a natural product hardwickiic acid isolated from plant Croton californicus with antinociceptive effect, and vixotrigine (also known as raxatrigine), which is a state- and use-dependent Nav1.7 blocker under clinical investigation for ameliorating trigeminal pain and neuropathic pain42,43,44. Our present structural studies, along with previous reports, reveal that the PD is a versatile receptor for ligands with distinct chemical formulas and structures.
Results
Cryo-EM analysis of Nav1.7 with different drugs
We started with more drugs than the abovementioned ones, including a local anesthetic lidocaine, an analgesic under development funapide (XEN402)45, a pain killer candidate DSP-22308, and a selective Nav1.7 inhibitor PF-05089771 that targets VSDIV46,47. To validate the function of some purchased antagonists, we performed preliminary whole-cell patch-clamp electrophysiological recordings in HEK293T cells (Supplementary Figs. 1–2 and Supplementary Tables 1–2). Based on these characterizations, the antagonists were incubated with purified Nav1.7-β1-β2 complex for 1 h at a final concentration of at least 10-fold higher than their measured IC50 values before cryo-sample preparation.
Following our reported cryo-EM data acquisition and processing protocols14,33, we obtained ten 3D EM reconstructions for the Nav1.7-β1-β2 complex treated with different small molecules at overall resolutions of 2.6−3.2 Å (Supplementary Figs. 3–4, Supplementary Figs. 8–10 and Supplementary Table 3). Cross comparison of 3D EM maps immediately reveals unambiguously resolved densities for bupivacaine (BPV), lacosamide (LCM), carbamazepine (CBZ), vinpocetine (VPC), hardwickiic acid (HDA), vixotrigine (VXT), and PF-05089771. However, no density is found for lidocaine, and the local resolution is insufficient to unambiguously assign funapide or DSP-2230, even when these compounds were added throughout the entire purification procedure during repeated attempt to resolve the ligands (Supplementary Table 5).
The lack of density for lidocaine and limited resolutions for funapide and DSP-2230 may result from various reasons. Detergents, which were applied at high concentrations, may interfere with the drug binding. In fact, we sought to solve the structure of human Nav1.5 bound to lidocaine with multiple attempts, but never observed any ligand density. It is thus not surprising that no lidocaine is found in the EM map for Nav1.7 either. Structures of both Nav1.5 and Nav1.7 display similar inactivated state. It is also possible that some ligands may prefer different conformations that are yet to be resolved. In the following, we will discuss the well resolved ligands only.
Recognition of PF-05089771 by Nav1.7-VSDIV was previously examined on a NavAb chimera where the extracellular half of the VSD is replaced by the counterpart in Nav1.7-VSDIV47. Our incentive to solve the structure of PF-05089771 bound to wild-type (WT) human Nav1.7 was to verify if the chimeric VSD can faithfully recapitulate all binding details. The binding mode of PF-05089771 in the intact Nav1.7-VSDIV is remarkably similar to the chimeric one (Supplementary Fig. 5). As coordination details and the molecular basis for the subtype-specificity have been thoroughly discussed by Ahuja and co-workers, we will not elaborate on this one. In the following, we will focus on drug binding to the PD.
A common site for BPV, LCM, and CBZ
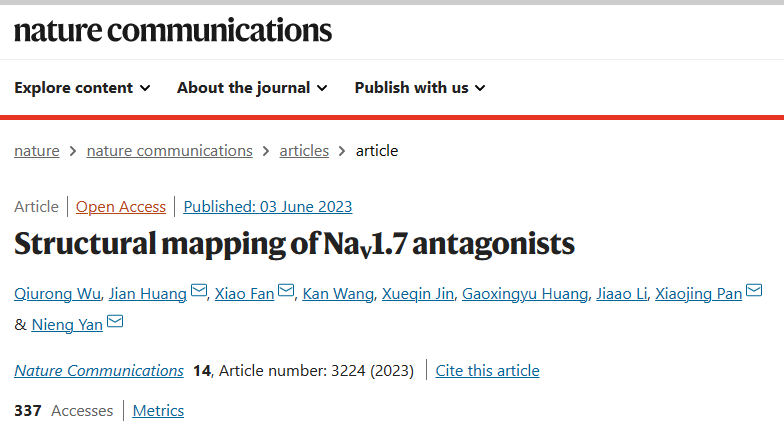
In the 3D EM map of all WT human Nav channels, the intracellular gate is penetrated by a linear density that likely belongs to the detergent molecule glyco-diosgenin (GDN) or digitonin. In the maps of Nav1.7 treated with BPV, LCM, and CBZ, the linear density is replaced by shorter ones with distinct features that respectively match the three drugs (Fig. 1a). Instead of wedging through the gate, these densities block it from the intracellular side. Therefore, BPV, LCM, and CBZ occupy the same site beneath the intracellular gate, which we name as site BIG. Unexpectedly, LCM has a second binding pose that directly plugs into the selectivity filter from the central cavity (Fig. 1b, c). We will discuss the second site in a later session. For illustration simplicity, we will name LCM as LCM-1 and LCM-2 in site BIG and the central cavity, respectively.
Fig. 1: BPV, LCM, and CBZ bind to the same site beneath the intracellular gate (site BIG).
a Distinct densities for the three drugs at site BIG. From left to right, the chemical structures (ball and sticks) and corresponding densities (semi-transparent cloud) are shown for GDN (Nav1.7-apo, PDB code: 7W9K), bupivacaine (BPV, brown), lacosamide (LCM-1, grey), and carbamazepine (CBZ, purple). The densities for the small molecules are presented at similar levels in Chimera, 5 σ for GDN, 4 σ for BPV, 4 σ for LCM-1, and 4.5 σ for CBZ. For visual clarity, only part of the S6I and S6III segments are shown. b The three drugs overlay at Site BIG. A bottom view (left) and a side view (right) of the superimposed pore domain (PD, domain colored) with bound drugs (BPV, LCM, and CBZ) are shown. The same color scheme for the four repeats is applied throughout the manuscript. c Drugs at site BIG directly block the intracellular gate. Shown here is a cross-section view of ligand-bound Nav1.7 PD in an electrostatic surface representation. Two distinct binding poses are observed for LCM (grey spheres), one at site BIG (LCM-1) and the other below the SF (LCM-2). d Further contracted intracellular gate in the presence of the site BIG-binding drugs. Left: Structural comparison of Nav1.7-BPV and Nav1.7-apo (silver). Middle: The pore radii of Nav1.7 bound to different ligands are calculated in HOLE and tabulated. Right: The α→ π transition of S6IV and rearrangement of the gating residues, such as Leu398, Leu964, Ile1457 and Ile1756 (shown as ball and sticks), in the presence of BPV lead to gate contraction. LCM and CBZ-bound structures display nearly identical conformations to that of Nav1.7-BPV.
Analysis of the ion permeation path using HOLE48 shows that the intracellular gate is substantially narrowed (Fig. 1d). The radius of the constriction site falls to ~1 Å, which is more than 1 Å shorter than that in the apo channel (PDB code: 7W9K)14. Gate contraction results from an α→π transition in the middle of the S6IV segment, in addition to minor inward movements of S6II and S6III (Fig. 1d). Similar conformational changes have been observed for Nav1.7 upon binding to the peptide toxins Protoxin-II (ProTx-II) or Huwentoxin-IV (HWTX-IV)14. ProTx-II binds to the extracellular side of VSDII and VSDIV, which are respectively known as Site 4 and Site 3 for neurotoxin binding to Nav channels; HWTX-IV recognizes Site 315, 49,50. It is interesting that peptide toxins and small molecule drugs that bind to unrelated sites lead to similar gate rearrangement.
In apo-Nav1.7, the intracellular gate is of an oval contour constituted by Leu398, Leu960, Phe963, Ile1453, Val1752, and Tyr1755 from the four S6 segments. In the presence of the drugs BPV, LCM, or CBZ, the three S6 segments other than S6I all slightly move toward the center in addition to the α→π transition of S6IV. Consequently, four gating residues, Leu398, Leu964, Ile1457, and Ile1756, move closer to tighten the gate (Fig. 1d). It is noted that Leu964, Ile1457, and Ile1756 are all engaged in the coordination of the three drugs (Fig. 2). Such conformational shifts are thus coupled to site BIG formation.
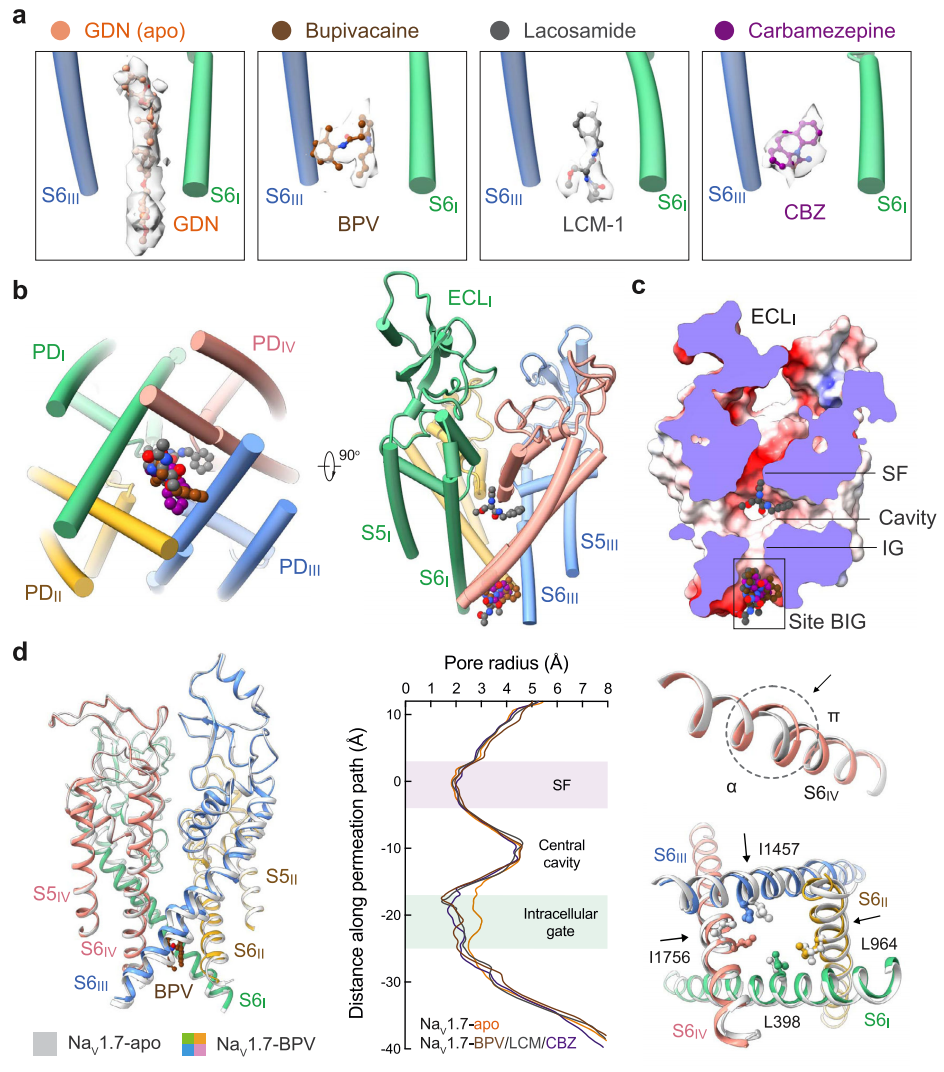
Fig. 2: Drug coordination at the BIG site.
Binding details of (a) BPV, (b) LCM-1, and (c) CBZ at site BIG are presented in identical side (top) and intracellular views (bottom). The drugs are shown as ball and sticks, and surrounding residues are shown as sticks. The potential hydrogen bonds are indicated with red dashed lines.
Site BIG
Site BIG is constituted by the cytosolic residues on the S6 tetrahelical bundle that are adjacent to the gating residues. Although the three drugs completely overlap in site BIG, the coordinating details vary. BPV is primarily surrounded by hydrophobic residues, including Leu964, Leu967, Leu968, and Phe971 from S6II, Ile1457 from S6III, and Ile1756 and Leu1760 from S6IV (Fig. 2a). The 3,5-dimethylphenyl and the piperidine rings of BPV, linked by an amide group, align on a similar plane against the BIG site, leaving the butyl tail pointing to the cytoplasmic side.
The purchased BPV is a racemate of dextro-(R)- and levo-(S)-bupivacaine. To investigate if BPV binding has racemic specificity, we performed in-silico docking simulations. The aromatic ring and the piperidine basic nitrogen of these two isomers largely overlap in site BIG. The primary difference in the binding poses of the isomers is the opposite orientations of the butyl tail (Supplementary Fig. 6). Therefore, both racemic isomers of BPV can be recognized at site BIG.
LCM-1 and CBZ occupy the same cavity of site BIG as BPV, but form additional hydrogen bonds (H-bonds) (Fig. 2b, c). The amide nitrogen from the central amide group of LCM-1 is H-bonded with the carboxyl group of Glu406 on S6I (Fig. 2b), and the carbonyl oxygen of CBZ receives an H-bond from Asn1461 on S6III (Fig. 2c). The three clinical drugs share a conserved binding mode, wherein the aromatic rings project into the center of the pore, leaving the flexible tail to the intracellular side. Drugs with the same chemical skeleton may share a similar binding pose.
LCM-2 directly blocks the SF
The central cavity of voltage-gated ion channels (VGICs) is a versatile platform for various chemicals. Eukaryotic Nav and Cav channels comprise four non-identical repeats, thereby offering more docking sites than homo-tetrameric VGICs.
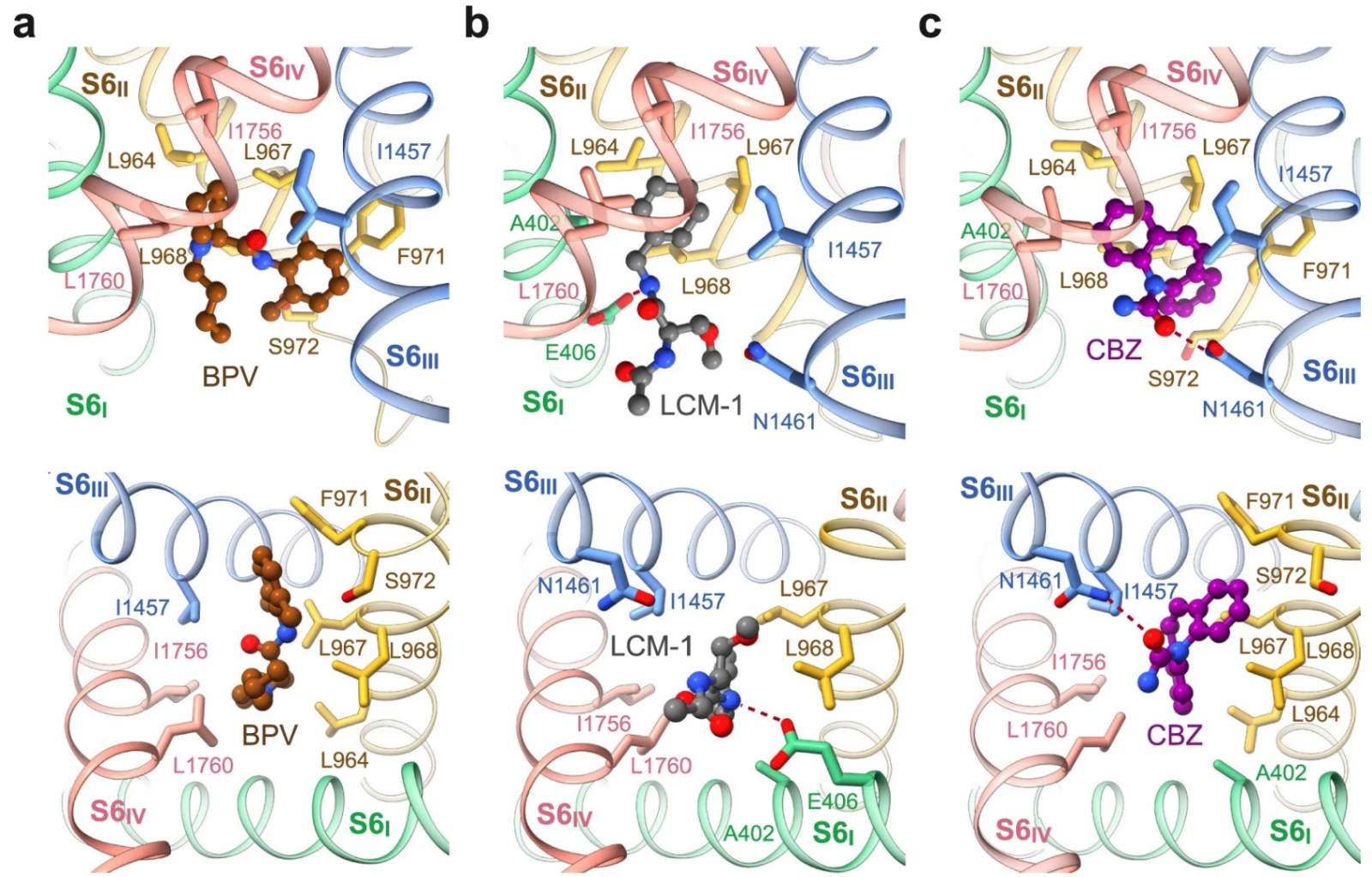
Beneath the ceiling of the central cavity, the Y-shaped LCM-2 is positioned off the central axis, closer to repeats I and IV (Fig. 3a, b). The benzyl ring is embedded in a pocket enclosed by residues Ser1697, Ile1744, Phe1748, Val1751, and Val1752. The two amides are H-bonded to the backbone carbonyl group of Thr1696 on the P1 loop of repeat IV, forming a stable anchor for LCM-2. The carbonyl oxygen from the acetylamino moiety interacts with Lys1406, which is one constituent of the DEKA motif on repeat III, and the amide nitrogen engages in an H-bond with Gln360, which is stabilized by the side chain hydroxyl group of Ser390 (Fig. 3c). The extensive H-bond network may account for the stronger density of LCM-2 than that of LCM-1 (Figs. 1a, 3a).
Fig. 3: Lacosamide binds to two distinct sites.
a Accommodation of lacosamide at the second binding site in the central cavity. The contour level for LCM-2 in the left panel is 5 σ. b A cut-open electrostatic surface of Nav1.7-LCM-2 viewed from the extracellular side. c Coordination of LCM-2 in the central cavity. Surrounding residues are shown as sticks. The potential hydrogen bonds are indicated with red dashed lines.
Vinpocetine and hardwickiic acid bind to the III–IV fenestration
The III–IV fenestration in the L-type Cav channels is a well-known site for dihydropyridine compounds13, 51,52. The corresponding site in Nav channels has been suggested to be targeted by local anesthetics53,54,55,56,57. Although no density for lidocaine is found around this site, densities for VPC and HDA were resolved40,58. A battery of hydrophobic residues on S6III and S6IV are engaged in accommodating VPC or HDA, including Thr1404 on P1III, Trp1332 on S5III, Thr1448, Leu1449, and Phe1452 on S6III, and Ser1697, Ile1744 and Phe1748 in repeat IV (Fig. 4a–c). Notably, the binding pose for VPC, which extends deeper into the central cavity, only partially overlaps with that of HDA in the III–IV fenestration (Fig. 4a).
Vixotrigine penetrates the IV–I fenestration
A stretch of elongated density is observed traversing the IV-I fenestration in VXT-treated Nav1.7. Linear density with similar pose is often found to belong to a lipid tail in apo-Nav channels. Fortunately, the bulges that are characteristic of the three ring moieties facilitated model building of VXT (Fig. 4a, b). VXT is held by a number of hydrophobic residues on S6I and S6IV, including Phe1692, Thr1695 and Thr1696 on P1IV, Ile386, Phe387 and Phe391 on S6I, and Val1751 and Tyr1755 on S6IV. The amido group from pyrrolidine moiety and carbonyl oxygen are H-bonded to the phenolic hydroxyl group of Tyr1755 on S6IV, forming a stable anchor for VXT (Fig. 4c).
Unlike VPC, the majority of which is within the cavity (Fig. 4b), VXT tunnels through the IV-I fenestration, with its acylamino moiety below the SF and its benzyl ring contacting the lipid bilayer (Fig. 4a, b). It is noted that the IV-I fenestration is sensitive to the α→π transition of S6IV (Fig. 4d). In the π form, the fenestration is gone. Such binding pose thus immediately provides a molecular interpretation for the dependence on channel states for VXT binding.
Discussion
A structural map for drug binding sites on Nav channels
Nav channels are well-known targets for natural toxins and clinically applied drugs. Seven sites (Site 1 – Site 7) have been mapped to the primary sequence of Nav channels based on decades of rigorous functional characterizations4 (Fig. 5a). With the growing number of high-resolution structures of Nav channels in the presence of toxins and drugs, the conventional definition of binding sites may no longer be able to reflect the diversity and complexity of the drug binding sites, particularly those on the PD. Hereby we map the known ligand binding sites to the resolved structures of Nav channels and propose a letter-coded nomenclature system to depict these druggable sites in the 3D space.
Resource linked:https://www.nature.com/articles/s41467-023-38942-3
Copyright© VastPro (Zhejiang) Pharmaceutical Co.,Ltd. ChinaChemNet Toocle Copyright Notice
4th Floor, Building 9B, 100 Haike Road, Pudong New Area District, Shanghai, 201210 P. R, China.
Tel: +86-21-20608178 Fax: +86-21-20608171 Contact us


