Detail
Source:https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00910
Baichen Xiong, Yuanyuan Wang, Ying Chen, Shuaishuai Xing, Qinghong Liao, Yao Chen, Qi Li*, Wei Li*, and Haopeng Sun*
Abstract
In the development of central nervous system (CNS) drugs, the blood–brain barrier (BBB) restricts many drugs from entering the brain to exert therapeutic effects. Although many novel delivery methods of large molecule drugs have been designed to assist transport, small molecule drugs account for the vast majority of the CNS drugs used clinically. From this perspective, we review studies from the past five years that have sought to modify small molecules to increase brain exposure. Medicinal chemists make it easier for small molecules to cross the BBB by improving diffusion, reducing efflux, and activating carrier transporters. On the basis of their excellent work, we summarize strategies for structural modification of small molecules to improve BBB penetration. These strategies are expected to provide a reference for the future development of small molecule CNS drugs.
1. Introduction
Central nervous system (CNS) function impairments and disorders [e.g., Alzheimer’s disease (AD), Parkinson’s disease (PD), stroke, and brain cancer] are widely considered extraordinarily severe pathological conditions. (1,2) According to the analysis of the Global Burden of Diseases, Injuries, and Risk Factors Study (GBD), neurological disorders are the leading cause of disability-adjusted life years (DALYs) and the second leading cause of death. (3) In particular, more than 50 million people suffer from AD globally and more than 10 million people worldwide live with PD. (4,5) Additionally, other CNS disorders, such as stroke and brain cancer, are also fatal diseases and cause a huge social and economic burden. (6,7) For example, malignant brain tumors have an extremely poor prognosis (three-year survival rate of 3–5%). (8) CNS disorders represent one of the largest markets for the development of new treatments. (9) In 2018, the global market value was $35.5 billion and is expected to continue growing because of the apparent unmet medical need. (10)
Treating CNS diseases requires drugs to be delivered into the brain in sufficient quantities to achieve therapeutic effects. (11) The major obstacle is the selective permeability of the blood–brain barrier (BBB), which prevents hydrophilic substances, charged molecules, proteins, and peptides from entering the brain. (12) This physiological hurdle of the BBB effectively blocks >98% of small molecule drugs and almost 100% of large molecule drugs, such as recombinant proteins and monoclonal antibodies, exerting therapeutic effects in the brain. (13) In fact, among the novel therapeutics approved by the Food and Drug Administration for the prevention, detection, or treatment of CNS diseases during 2016–2020, most of them were small molecule drugs as new molecule entities (NMEs), while biologics license applications (BLAs), including antibody-based and oligonucleotide-based therapeutics, accounted for only a small portion. (14,15)
The BBB is a permeable barrier formed between the blood and brain that plays the role of maintaining homeostasis of the extracellular microenvironment in the neural tissue of the brain as well as protecting against neurotoxic compounds. (16,17) More than 100 years ago, Ehrlich found that some dyes injected into the vascular system were excluded from the brain. (18) To date, the structure, morphology, and signaling pathways of the BBB have been extensively studied. (19) The BBB is formed by brain microvascular endothelial cells (BMECs), pericytes, and astrocytic end feet. (20) BMECs act as building blocks of the BBB and differ from other endothelial cells, containing a higher concentration of mitochondria and an absence of fenestrations. (21,22) In addition, BMECs impede the entry of most molecules, with the exception of small and lipophilic molecules. (23) In addition to passive penetrations, efflux and carrier-mediated transcytosis (CMT) are also common mechanisms of exchange (Figure 1). BMECs contain efflux transporters for breast cancer resistance protein (BCRP) and multiple drug resistance 1 (MDR1) that is also termed P-glycoprotein (P-gp). They serve to transfer the substrate compound from the brain. (24,25) CMT works by transporting proteins on BMECs that specifically recognize and deliver brain-necessary substances, such as glucose, amino acids, purine bases, and nucleosides. (26)
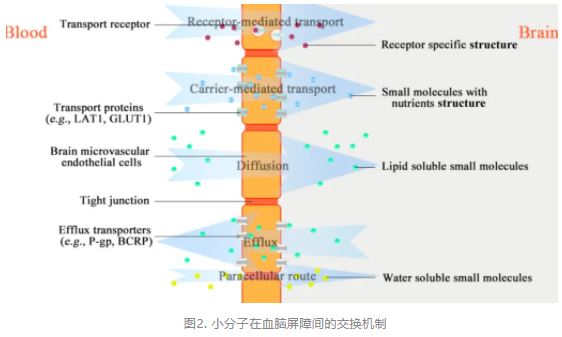
Figure 1. Mechanisms of exchange of small molecules across the BBB.
Many efforts have been made, and several brain drug delivery approaches have been developed. Intracerebral administration, intranasal drug delivery, and nanoparticle (NP)-mediated drug delivery are both promising approaches for the delivery of therapeutically active compounds directly into the CNS. (27−30) From this perspective, excellent works of medicinal chemists reported in the past five years were reviewed. Structural modification to influence BBB penetration by modulating either passive diffusion or active transport could provide useful strategies for improving BBB penetrability. The development of research in this area compared to five years ago and the possible trends in the future are also worth discussing. (31,32) We give our own view about this herein.
2. Important Tools and Parameters in Brain Penetration Evaluation
To provide guidelines for drug design, a variety of methods and models for assessing the potential of small molecules to permeate the BBB have been developed. Through every step of medicinal chemical work, in silico simulation based on reliable data and modeling approaches rationally directs structural modification. In vitro methods can conveniently predict BBB penetration and are usually used for choosing optimized compounds to carry out in vivo experiments, which provide the most reliable reference information for testing and validating in silico models. (33,34) Some important tools and parameters for CNS drug discovery are discussed in this section.
2.1. In Silico Simulation Based on Physicochemical Properties
As early as 1997, Lipinski described experimental and computational approaches to estimate solubility and permeability in discovery and development settings, with Lipinski’s rule of five (RO5) proposed in addition. (35) This method of predicting permeability based on physicochemical properties has been continuously developed and used in studies to determine the potential druggability of compounds. (36−38) In CNS drug research, physicochemical properties are also critical for guiding drug design, lead optimization, and candidate selection. Published reports have suggested that some physicochemical characteristics are decisive with respect to the ability of a molecule to pass the BBB. (39) The specific meaning and preferred ranges are listed in Table 1.
| property | comments | suggested rangesa |
|---|---|---|
| cLogP | octanol–water partition coefficient | 2 < cLogP < 4 (40) |
| cLogD (pH 7.4) | octanol–water cLogP at pH 7.4 | 2 < cLogD < 3 (41,42) |
| HBD | hydrogen bond donor, estimated number of hydrogen bonds that would be donated to the solvent water | HBD < 3 (43) |
| MW | molecular weight | MW < 450 (42,44) |
| pKa | acidity coefficient | 6 < pKa < 10.5 (45) |
| tPSA | topological polar surface area, the sum of surfaces of polar atoms |
aSuggested physicochemical property ranges of small molecule compounds that can penetrate the BBB.
In 2010, Wager at Pfizer developed a multiparameter optimization (MPO) algorithm that combined six physicochemical parameters. (47) In recent years, CNS MPO desirability tools have been widely used in prospectively designing molecules. (48) Utilizing a flexible, multiple-parameter algorithm rather than single parameters could more comprehensively estimate the potential of small molecules to permeate the BBB. In other words, the CNS MPO method can balance multiple variables while avoiding hard cutoffs, which is beneficial for expanding the design space for CNS drugs and taking into account absorption, distribution, metabolism, and excretion (ADME) attributes together with BBB penetrability. (49) In addition to the CNS MPO algorithm, other scoring systems for the prediction of BBB penetration have been successively published, such as the technically enhanced multiparameter optimization (TEMPO) score of Ghose et al. and the BBB score of Weaver et al. (50,51) Even though the accuracy of these new algorithms was suggested to be better than that of the CNS MPO, they could be used only as a reference. Excellent CNS MPO scores are sometimes relevant to low brain exposure. (52)
2.2. In Vitro Methods for Predicting BBB Penetration
In vitro methods are relatively accurate compared with in silico models and convenient compared to labor-intensive in vivo experiments. Therefore, they have the advantage of high-throughput application and are usually used to predict BBB penetration and determine drug candidates. (53) The common in vitro tools for CNS drug discovery are discussed below.
The parallel artificial membrane permeability assay (PAMPA), developed by Kansy et al. in 1998, is a permeation model for investigating passive absorption processes. (54) The PAMPA is used to evaluate drug permeability across various biological membrane systems. Tested compounds diffuse from the donor well into the acceptor compartment through the artificial membrane monolayer. In the PAMPA–BBB assay, the artificial membrane that mimics the BBB is produced by an extract from a porcine brain containing specific phospholipids. (55) Usually, some commercial drugs with reported values are chosen as references to verify a linear correlation. (56,57) The effective permeability (Pe) is calculated to represent the permeability of the samples. PAMPA has the advantages of low cost and reproducibility. However, the limitation of this technology is that the artificial membrane is not a bilayer lipid structure. Moreover, a lack of transporters leads to an inability to reflect efflux and CMT, which causes deviations between the measurement results and the actual situation. (58)
Cell-based monolayer transport assays apply polarized monolayers formed by cells to build in vitro BBB models, including non-cerebral origin cell lines such as modified Madin-Darby canine kidney cells (MDCK), human colon carcinoma cells (Caco-2), and immortalized brain endothelial cell lines such as rat endothelial cells (RBE4). (59,60) Modified MDCK cell lines, such as MDCK-MDR1 and MDCK-BCRP, are extensively used in drug discovery to evaluate transporter-mediated efflux. (61) P-gp (MDR1) and BCRP are two important efflux drug transporters that are strongly expressed at the BBB. The efflux ratio (ER) is a significant parameter that influences the brain penetration of drugs. Differentiated MDCK cells were seeded in 96-well transwell plates at a sufficiently high density to form a polarized monolayer. The transport of the test compounds from apical to basolateral (A-B) and from basolateral to apical (B-A) was determined. Samples from the apical and basolateral sides were collected and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) to calculate the apparent permeability coefficient (Papp) and ER. Most CNS drugs have high passive permeability with Papp values of >15 × 10–6 cm/s. “ER < 2.5” suggests that the compound is not a substrate for the P-gp or BCRP efflux transporters. (62)
With technological advancements in BBB modeling in recent years, highly integrative and quasi-physiological in vitro models have been established. (63,64) Based on microfluidic systems or the application of stem cells and three-dimensional printing technologies, these in vitro models have a better correlation with in vivo values. However, higher costs hamper the development for widespread use. (65)
2.3. In Vivo Experiments for Measuring the Drug Ratio in the Brain
In vivo approaches for assessing the CNS penetration of compounds in the brain have better accuracy and reliability. Commonly used methods are Cbrain/Cplasma determination, microdialysis, brain exposure assessment (BEA), etc.
Cbrain/Cplasma has a wide range of applications as a relative indicator of brain distribution. In addition, it appears to be the most frequently used method, because of its experimental simplicity. Blood and brain samples are collected at designated postdose time points. The test drug concentrations were analyzed using LC-MS/MS following well-established extraction procedures. (66)Cbrain/Cplasma values of less than 0.1 are interpreted as significant limitations to free BBB penetration. (67)
In recent years, the unbound drug concentration (Cu) based on the free drug hypothesis has gradually received more attention. The hypothesis states that only Cu is responsible for pharmacological activity in the brain. The fraction unbound (fu) is proposed to define the free drug available in tissues. (68) According to the unbound drug concentration in the plasma [Cu,p = total drug concentration in the plasma (Cp) × fu,p] and the brain [Cu,b = total drug concentration in the brain (Cb) × fu,b] and the corresponding unbound brain-to-plasma partition ratio (Kp,uu = AUCu,b/AUCu,p), new experiments for measuring the drug ratio in the brain have been established. (69) A Kp,uu of 0.3 was used as a cutoff to classify compounds as brain penetrant or not brain penetrant in vivo. (8)
Microdialysis is a profitable technique for measuring Cu,b. The hollow fiber dialysis membrane is connected with a probe inserted into the brain parenchyma of the animal. The microdialysis probe is continuously perfused with isotonic solution, and small molecules in the brain extracellular fluid are in equilibrium with the perfusate. Lastly, the effluent is collected and analyzed. (70,71)
Microdialysis is technically challenging and consumes resource. BEA was developed in early drug discovery to evaluate compound CNS distribution using Kp,uu as the index. Briefly, the assessment is based on a combination of Cp and Cb obtained in vivo, as well as fu,p and fu,b determined in vitro. (72) The total drug concentration can be easily determined by the method described above. For the in vitro part, many methodologies have been developed for determining the extent of plasma or brain protein binding of drugs. For example, equilibrium dialysis is one of the most utilized methods. The tested compounds added to plasma or brain homogenate were placed in the sample chamber and reached steady state in 6 h with the buffer placed in the receiving chamber. The fu is calculated as the ratio of the drug concentration in the receiving chamber and the total drug concentration. (73)
3. Strategies for Enhancing the BBB Permeability or Reducing the Efflux Ratio
In the past five years, adjusting the passive diffusion has remained the most effective means for modifying the structure to increase the CNS penetrability. Physicochemical properties are the key points for improving the BBB permeability of drugs. Compared to previous studies, structural modifications in recent works were based on multiple parameters rather than single parameters. It avoids sacrificing target activity but also takes into account both CNS penetration and potential druggability.
Blood–brain barrier penetration depends on the lipophilicity of the compound in most cases. However, not surprisingly, it is noteworthy that discrepancies often occur between lipophilicity and brain exposure to many compounds because of the existence of multiple drug transport mechanisms through the BBB. Many drugs with high lipophilicity have a very low concentration in the brain due to the active efflux mechanism of the BBB. Both P-gp (MDR1) and BCRP are significant drug efflux transporters. (74) They were originally identified as a crucial reason for multidrug resistance in the treatment of certain cancers and have been found to be expressed at the luminal side of brain capillary endothelial cells forming the BBB. (75) In drug exploration, MDCK-MDR1 and MDCK-BCRP are widely used to evaluate transporter-mediated efflux. ER is calculated as Papp (B–A)/Papp (A–B). When ER > 2.5, thus the experimental results imply a high possibility of efflux.
The most intensively studied transporter is P-gp, which is strongly expressed in the BBB and is the reason why many drugs cannot cross the brain. However, there is no strictly defined structure that is the substrate of the efflux transporter because efflux transporters have multiple binding sites and complex mechanisms of substrate recognition and transport. Therefore, specific structure–activity models of efflux transporters, including P-gp, have proven to be difficult to build. (76) Fortunately, there is a correlation between the binding tendency of the P-gp substrate and the physicochemical properties of the compound. The good information is that property changes in passive diffusion also likely decrease the affinity of binding to P-gp. Eliminating the HBD, reducing the molecular size, replacing electronegative atoms such as O with S or CH2, and introducing a constrained conformation are also examples of optimizing a P-gp inhibitor. (77)
The following are some common strategies for enhancing the BBB permeability or reducing the efflux ratio, with several recent works illustrated as references.
3.1. Increasing Lipophilicity
Lipophilicity is defined as the partition coefficient of an un-ionized compound in two immiscible phases (such as n-octanol and water/buffer) at equilibrium, revealing the extent of a compound’s solubility in lipids, oil, or nonpolar solvents. (78) Lipophilicity is the first parameter found to be closely related to CNS penetration. However, as one of the most crucial physicochemical properties and one of the most frequently analyzed parameters, lipophilicity is also a pivotal indicator of pharmacokinetic and dynamic information. An increasing lipophilicity also affects potential druggability due to changes in water solubility, metabolic half-life (t1/2), and clearance (CL). (79) Therefore, only a few works, mainly through improving lipophilicity, enhanced BBB permeability. The incorporation of fluorine is commonly used in structure optimization. (80) Although the lipophilicity of compounds does not always increase upon introduction of fluorine, strategic application of fluorine in specific structures could enhance lipophilicity while improving metabolic stability by occupying the site of oxidative metabolism. (81) Furthermore, the small size of fluorine allows it to be readily incorporated into a molecule to tune electronic properties without causing large disruptions in the steric environment. (82) Pettersson et al. examined a large compound data set and concluded that an increased MW due to the introduction of fluorine does not lead to increased MDR ER liability. (83)
For example, anaplastic lymphoma kinase (ALK) is an attractive therapeutic target for cancers that have ALK gene fusions or activating mutations. One of the extensively used ALK-inhibiting drugs, crizotinib (1), has been proven to have poor activity against CNS metastases of cancer due to the inability to cross the BBB. (84) Recently, Worms and colleagues published studies that sought to develop a fluoroethyl moiety addition. The region of analysis confirmed that lipophilicity increases brain permeability. The %ID/cc of [18F]fluoroethyl crizotinib ([18F]2) at 5 min after injection reached 6.6, while the value expected for a tracer confined to the vascular compartment was just 0.2%ID/g. The authors estimated that the therapeutic potential of 2 may exceed that of the lead compound, crizotinib (Figure 2). (85)
Figure 2
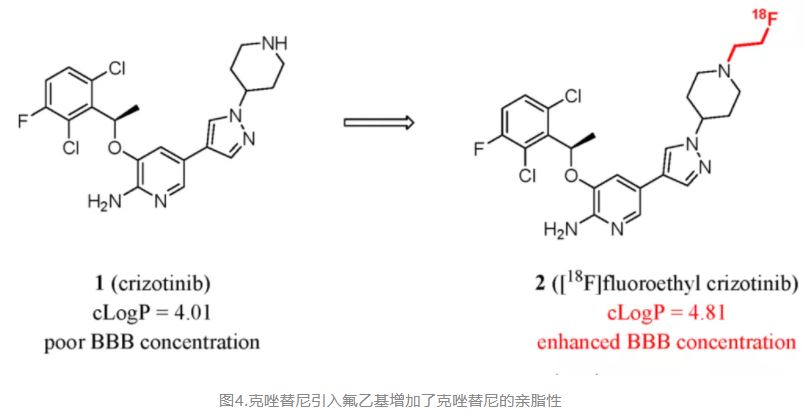
Figure 2. Crizotinib derivative as ALK-inhibiting drugs and positron emission tomography (PET) tracers: enhancing lipophilicity to enhance BBB penetration.
Brand et al. developed lead compound 3 (DDD85646) from the optimization of a high-throughput screening hit. Compound 3 has potent activity against N-myristoyltransferase (NMT), which represents a promising drug target for human African trypanosomiasis (HAT) caused by the parasitic protozoa Trypanosoma brucei. (86) However, 3 was not blood–brain barrier penetrant (Kp < 0.1). Wyatt and co-workers recently sought to design and discover a series of derivatives of compound 3 with improved BBB penetrability. The study reported that the low brain exposure was attributed to the high tPSA (89 Å2) and low lipophilicity (cLogP = 1.79). On the basis of this consideration, compound 4 was designed and synthesized. In the following modification, lipophilicity was further improved. Compound 5 showed remarkable inhibitory activities, good microsomal stability, and significant levels of brain penetration (Figure 3). (87)
Figure 3
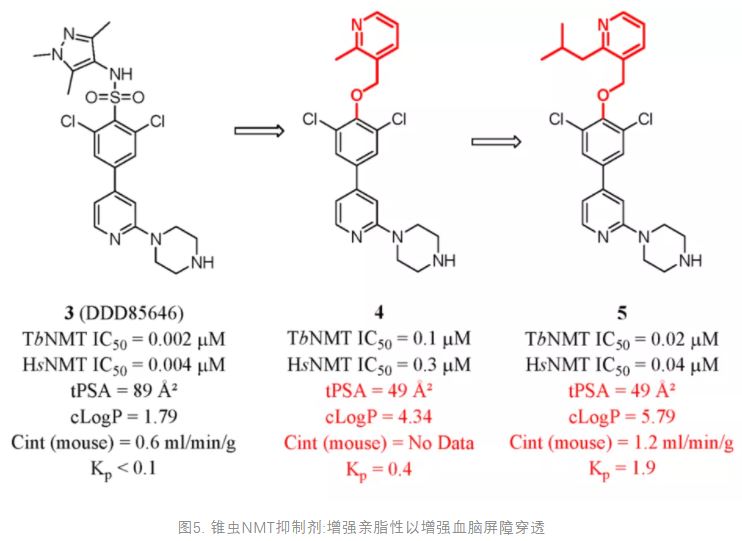
Figure 3. Trypanocidal NMT inhibitors: enhancing lipophilicity to enhance BBB penetration.
3.2. Reducing Hydrogen Bond Donor Capacity
RO5 defines the HBD of desirable drug candidates as <5, while CNS drugs usually have fewer HBDs (<3). In the recent research, reducing HBD capacity has been one of the most frequently utilized drug design strategies for improving BBB penetration.
For example, Sakai et al. replaced the 5-aminopyrimidine of 6 with 3-chloropyridazine to gain a fibroblast growth factor (FGF) receptor modulator 7 with improved brain exposure [total brain-to-plasma partition ratio (Kp) = AUCb/AUCp] and reduced risk of phospholipidosis. Modulator 7 may have the potential to imitate the biological activities of basic FGF, such as neuroprotective and cell proliferative activities, showing promise as a profitable agent for the treatment of neurodegenerative diseases (Figure 4). (88)
Figure 4
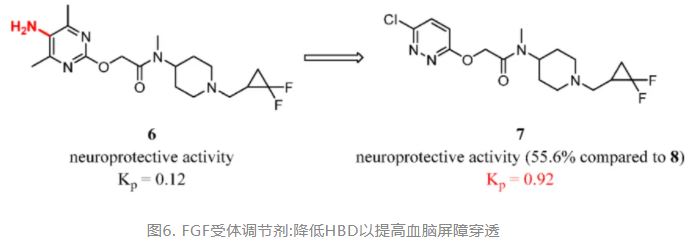
Figure 4. FGF receptor modulators: reducing HBD to improve BBB penetration. Kp was determined in rat by 1 mg/kg intravenous injection (iv).
Structure-based drug design has been widely used in medicinal chemistry research in recent years. Effective and highly rated successful structural modifications are based on virtual docking or co-crystal structures rather than random attempts. Using co-crystal structures for guidance, Fujimori and colleagues explored the removal of hydrogen bond acceptors (HBAs) and HBDs of compound 8 that did not form essential interactions with the ALK active site. As described above, ALK inhibitors have been identified as effective drugs for treating cancers that have ALK gene fusions. As expected, removing one of the HBDs to obtain compound 9 significantly inhibited MDR1 efflux, indicating a great improvement in brain exposure. When the oxygen atom on the morpholine ring was shielded by a methyl group, the MDR1 efflux ratio further decreased. The authors announced that 10 could be valuable as an in vivo tool for elucidating the role of ALK in CNS disorders such as cognitive impairment, anxiety, and depression (Figure 5). (89)
Figure 5
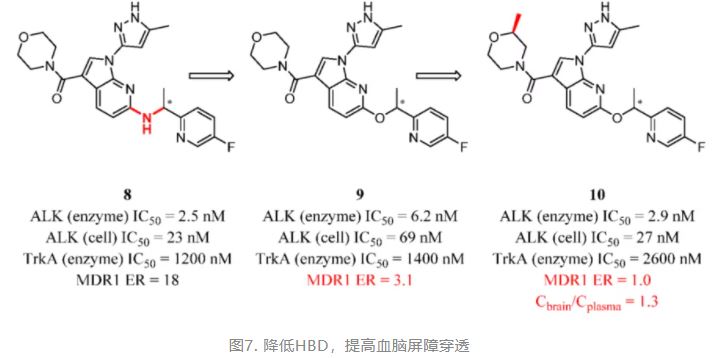
Figure 5. ALK inhibitors: reducing HBD to improve BBB penetration. Cbrain/Cplasma was determined in mice at 5 min after 100 mg/kg intraperitoneal injection (ip).
When the HBD in the structure is the key to maintaining the activity of the drug, building intramolecular hydrogen bonds is a good strategy for enhancing permeability. As reported by Dighe et al., intramolecular hydrogen bonds concealed the polar hydroxy and tertiary amine groups of compounds 11 and 12, which exhibited a permeability higher than that of 13 without intramolecular hydrogen bonding. Compounds 11 and 12 have both potent Aβ42 fibrillation inhibition and good membrane permeability and are regarded as prospective multifunctional drugs for AD (Figure 6). (90)
Figure 6
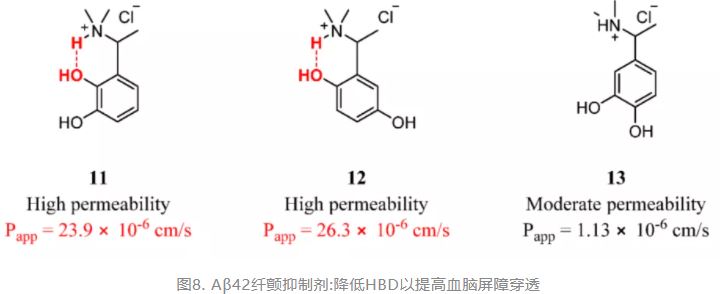
Figure 6. Aβ42 fibrillation inhibitors: reducing HBD to improve BBB penetration.
Reversing the strategy, which means an increased number of HBDs, could help develop highly peripheral selective drugs. IKur encoded by the KCNA5 (KV1.5) gene is important in atrial repolarization. The 5-phenyl-N-(pyridin-2-ylmethyl)-2-(pyrimidin-5-yl)quinazolin-4-amine 14 is a potent IKur current blocker that is a potentially safe agent for the maintenance of normal sinus rhythm. However, compound 14 with high brain exposure had the potential for CNS side effects. (91) After the transformation of the introduction of hydrogen bond donors, Finlay and co-workers discovered compound 15 with low brain exposure while maintaining the in vitro potency and selectivity profile of the IKur current blocker (Figure 7). (92)
Figure 7
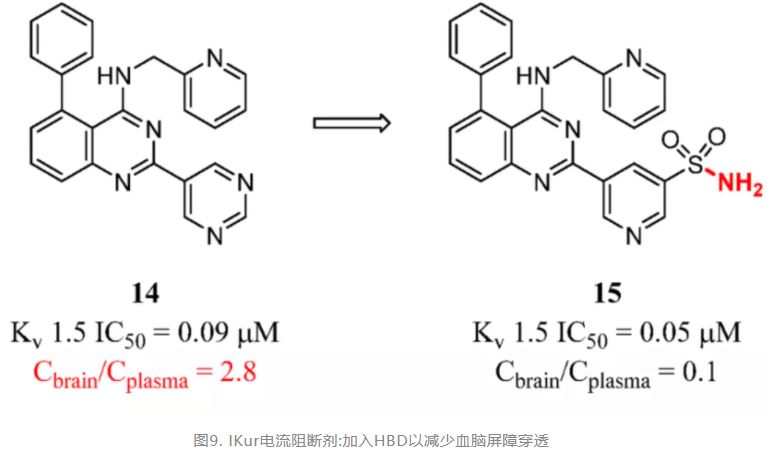
Figure 7. IKur current blockers: adding HBD to reduce BBB penetration. Cbrain/Cplasma was determined in mice at 10 min after 3 mg/kg (iv).
In the past five years, many medicinal chemists have used HBD removal to improve BBB penetration. The efforts are shown in the following pictures. Lindsley and co-workers replaced the primary sulfonamide moiety of a muscarinic acetylcholine receptor subtype 4 (M4) positive allosteric modulator (PAM) lead compound 16 and further displaced the amide linker. Compound 17 (VU6002703) stood out as a potent M4 PAM (hM4 EC50 = 600 nM) with excellent CNS drug potential (Kp,uu = 0.74) (Figure 8a). (93) Lindsley’s studies also reported their efforts to develop another M4 PAM scaffold based on a 6-fluoro-4-(piperidin-1-yl)quinoline-3-carbonitrile core. Compound 18 (VU0619856) represented an exciting new chemotype for the M4 field (hM4 EC50 = 897 nM) but showed little brain exposure. Additionally, using this method, removing the secondary amide H bond donor led to improved M4 PAM functional potency and enhanced CNS penetration (Figure 8b). (94) In addition, in an optimization of the pyrazolo[1,5-b]pyridazine scaffold for the treatment of human African trypanosomiasis by Pollastri and colleagues, the hydroxyl group hindered access of compound 20 to the brain. Compound 21 was fast acting and parasiticidal and showed good BBB penetration in the PK study (Figure 9). (95)
Figure 8
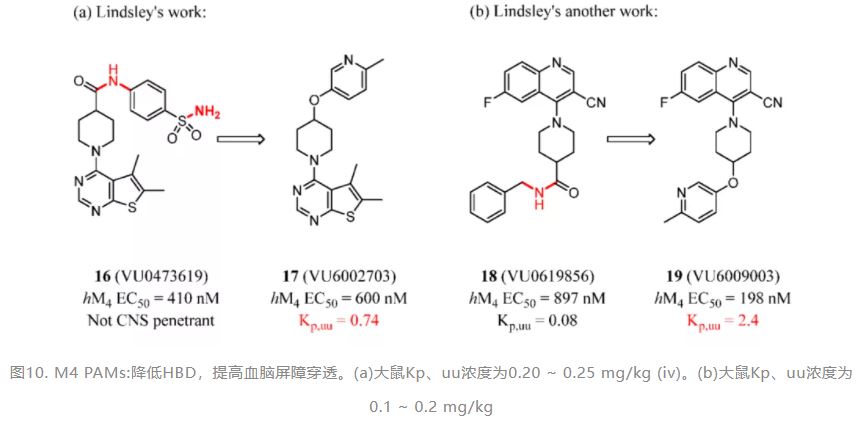
Figure 8. M4 PAMs: reducing HBD to improve BBB penetration. (a) Kp,uu was determined in rats by 0.20–0.25 mg/kg (iv). (b) Kp,uu was determined in rat by 0.1–0.2 mg/kg (iv).
Figure 9
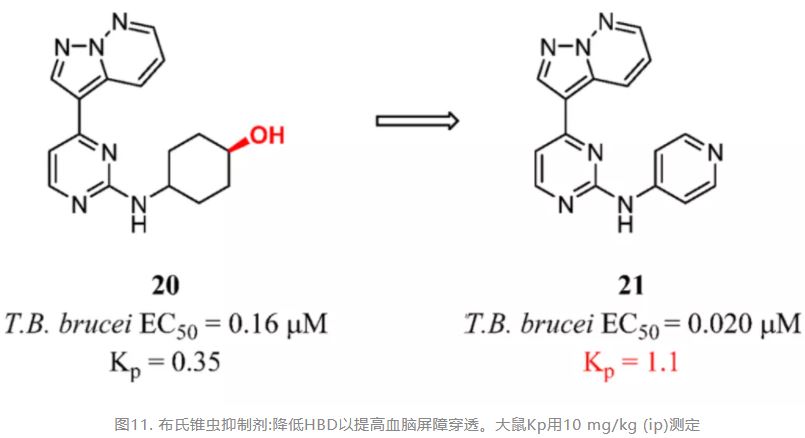
Figure 9. Trypanosoma brucei inhibitors: reducing HBD to improve BBB penetration. Kp was determined in rats by 10 mg/kg (ip).
3.3. Reducing tPSA
tPSA, a descriptor defined as the sum of surfaces of polar atoms in a molecule, has increasingly served as one of the most useful parameters for predicting molecular transport properties since it was disclosed by Ertl et al. in 2000. (96) tPSA is related to the number of polar atoms in the molecules and influences BBB permeability, drug metabolism, efflux, etc. (97) CNS drugs commonly have low tPSA values (tPSA < 90 Å2). In recent years, there have been some works to improve brain exposure around the tPSA.
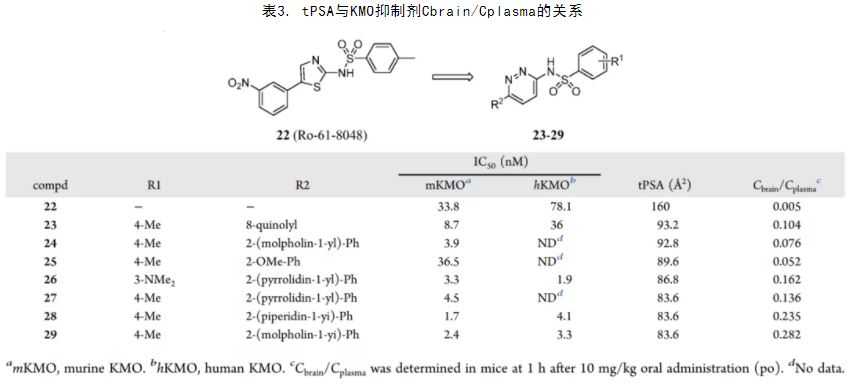
Kynurenine monooxygenase (KMO) is regarded as an ideal drug target for Huntington’s disease (HD). Inhibiting KMO is expected to reduce the toxic metabolites 3-hydroxykynurenine and quinolinic acid, which are involved in HD. 22 (Ro-61-8048) reported by Röver et al. is a KMO inhibitor with poor brain efficacy. (98) Isobe et al. proposed the reason as a high tPSA (tPSA = 160 Å2). Selected from their compound library, compound 26 with the same bis-heteroaryl sulfonamides showed superior inhibitory activity and tPSA (tPSA = 80.3 Å2). In the SAR exploration of compound 26, the relationship between tPSA and BBB penetration was clearly observed (Table 2). Compound 29 resulted in single nanomolar level efficacy and moderate brain exposure. Reducing tPSA is an effective strategy to improve BBB penetration. (99)
Table 2. tPSA–Cbrain/Cplasma Relationship of KMO Inhibitors
Zwicker et al. recently revealed studies toward developing potent inhibitors of Toxoplasma gondii cathepsin L (TgCPL), a viable target for the treatment of parasitic infections. Lead compound 30 was an inhibitor of human cathepsin L (HsCPL), which had a high tPSA (tPSA = 97.6 Å2). Guided by a structure-based design, investigators attempted to remove polar atoms not actively engaged by the enzyme to reduce overall tPSA. For example, the amide was transformed with the well-established isostere trifluoroethylamine. High potency against Toxoplasma and BBB penetrance of 31 suggested that bioisosterism was an outstanding modification method (Figure 10). (100)
Figure 10

Figure 10. CPL inhibitors: reducing tPSA to improve BBB penetration. Cbrain/Cplasma was determined in mice at 0.5 h after 10 mg/kg (ip).
3.4. Enhancing Rigidity
Central drugs usually have a strong rigidity, which manifests as a multiring system and a few rotatable bonds. According to the recommendation by Ghose et al., molecular flexibility can be represented by the number of linear chains outside rings and the number of rotatable bonds (NRB). The preferred ranges were two to four linear chains outside rings and one to four rotatable bonds. (41) Several druglike rigid fragments have been successfully and widely used. For example, benzazepine is a common fragment found in many CNS drugs, such as diazepam, galantamine, and lorcaserin (Figure 11). (101) The following are excellent investigations of adjusting molecular rigidity to affect BBB permeability.
Figure 11
Figure 11. CNS drugs with the benzazepin fragment.
Erlotinib (32) is one of the first-generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) widely utilized for the treatment of terminal non-small-cell lung cancer (NSCLC) patients with sensitive EGFR mutations. The epidermal growth factor receptor is mutated and amplified in nearly 60% of glioblastoma (GBM) tumors. However, all studies using generation EGFR TKIs failed to improve the outcomes of patients with GBM, because they do not cross the BBB in sufficient concentrations to achieve therapeutic consequences. Therefore, a novel EGFR TKI with high brain exposure has broad market and application potential. Nathanson and colleagues proposed that erlotinib’s poor brain penetration was attributed to flexible alkyl ether tails, including a large NRB (10) and high tPSA (75 Å2). On the basis of this hypothesis, they closed the flexible alkoxy chains to form a 1,4-dioxane ring fused onto the quinazoline scaffold. This modification led to 33, which contains a reduced NRB (10 to 2) and tPSA (75 to 56 Å2). Compared to erlotinib, compound 33 increased BBB penetration (Kp,uu = 0.491) by nearly 10-fold. After the introduction of haloalkyl substitutions, brain exposure of 34 (JCN037) further increased (Kp,uu = 1.30). At the same time, the novel and brain penetrant EGFR TKI 34 displayed potent activity against EGFR-altered primary GBM cells both in culture and in orthotopic xenografts (Figure 12). (102)
Figure 12

Figure 12. EGFR inhibitors: enhancing rigidity to improve BBB penetration. Kp,uu was determined in mice by 10 mg/kg (po).
Recently, Bruno and co-workers utilized the strategy of replacing iminoether by five-membered heterocycles bearing the same alkyl chains to obtain more information about pharmacophore features. Phosphodiesterase type 4D (PDE4D) is responsible for catalyzing the decomposition of cAMP, which is a key step in memory formation. Inhibiting PDE4 has been indicated to facilitate recognition in degenerative diseases such as AD. (103) In the docking and molecular dynamics simulation of lead compound 35 (GEBR-7b) and the crystal structure of PDE4D, the iminoether moieties played a significant role in forming a wide network of stable, direct, and water-mediated H bond interactions with the catalytic site of PDE4D. Heterocycle insertion reduced the molecular flexibility and gave fewer conformers. In the pharmacokinetic analysis, compound 36 was rapidly absorbed and reached the brain. Compared to that of 35, the BBB penetration of 36 obviously increased (Figure 13). (104)
Figure 13
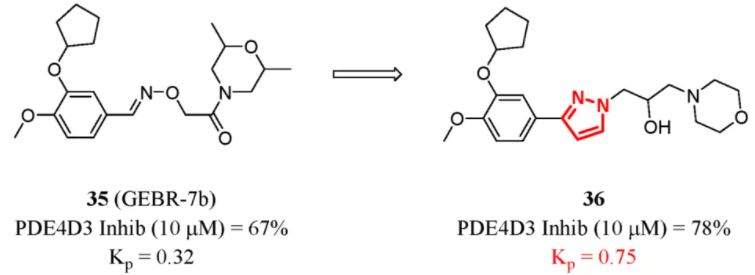
Figure 13. PDE4D inhibitors: enhancing rigidity to improve BBB penetration. Inhib (10 μM) means the inhibition rate in 10 μM. Kp was determined in mice by 10 mg/kg subcutaneous injection (ih).
3.5. Reducing pKa
Reducing pKa is also one of the most frequently utilized drug design strategies for improving BBB penetration. In addition, some of the studies discussed above proved it collectively. For example, as shown in Figure 2, the increase in brain exposure could also be attributed to the substitution of the N atom on piperazine, which leads to a decrease in basicity.
Furthermore, pKa has a significant correlation with ER. (46,105) Many investigators have noticed the obstruction of efflux on BBB penetration and attempted to solve it by reducing pKa.
In the work referenced in Figure 4, the original lead compound was 37 (Figure 14). As two similar structures, 6 was not a substrate for P-gp (ER = 1.1), while 37 was (ER = 6.3). The authors proposed that the reduction in basicity may contribute to the avoidance of P-gp recognition. (88)
Figure 14
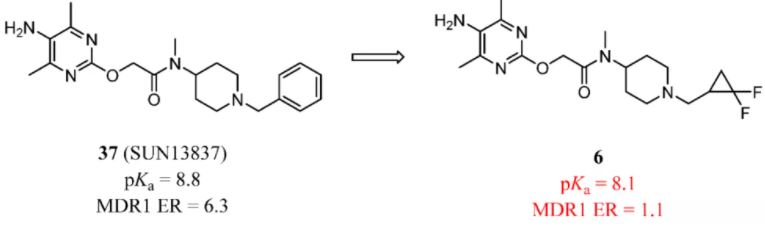
Figure 14. FGF receptor modulators. Reduction of basicity may contribute to the avoidance of P-gp recognition.
Koriyama and co-workers recently disclosed the development of β-site amyloid precursor protein cleaving enzyme 1 (BACE1) inhibitors that can block the rate-limiting step of synthesizing amyloid β peptides (Aβ). Aβ accumulation plays a critical role in the pathogenesis of AD. In addition, the inhibition effect offers the potential to delay or even prevent disease progression. (106) Compound 38 showed high P-gp efflux (ER = 52 in MDCK cells). Analysis of physicochemical features related to P-gp efflux indicated that a compound with a pKa of <8 would be less likely to be a P-gp substrate. The pKa for 39 was determined to be 9.0. On the basis of a rational design of the pKa-decreasing approach, thiazine-based BACE1 inhibitors 39 (JNJ-54861911, Atabecestat) with the most promising profile and decreased ER were advanced to further in vivo studies (Figure 15). (107)
Figure 15
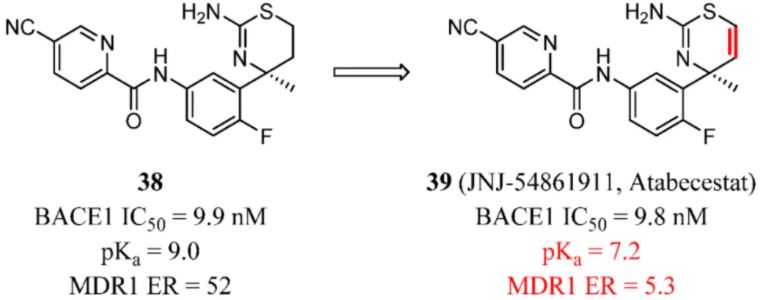
Figure 15. BACE1 inhibitors: reduce basicity to reduce MDR1 ER.
In the design of phosphodiesterase 10A (PDE10A) inhibitors, a dual substrate PDE that catalyzes the hydrolysis of cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP), two similar structures showed extraordinary differences in blood–brain distributions. Investigators assessed TgCPL inhibitors 40 and 41in vitro to determine whether they were potential substrates of P-gp. The high ER of compound 41 clarified its poor brain exposure in vivo. The authors attributed the shortcoming to the presence of a basic amine in 41 (Figure 16). (108)
Figure 16
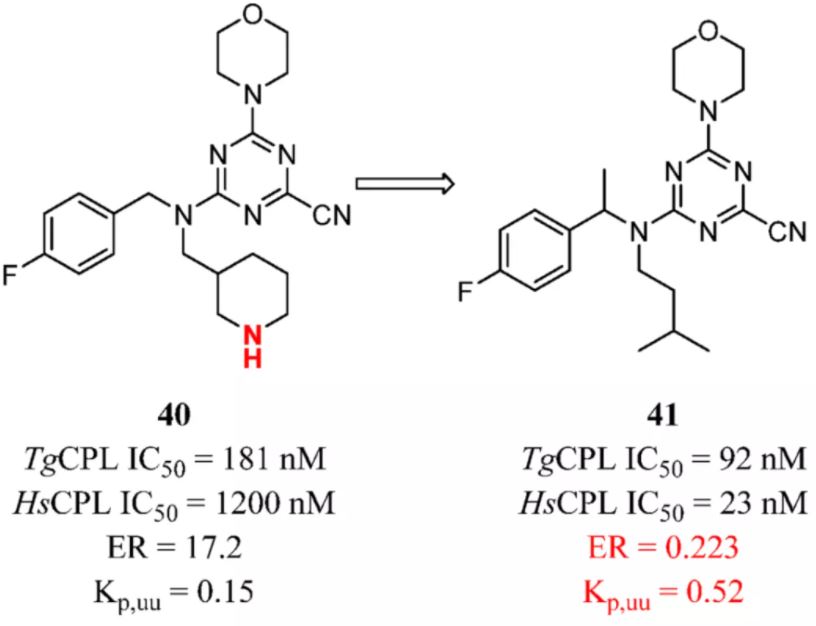
Figure 16. TgCPL inhibitors: reducing pKa to improve BBB penetration. Kp,uu was determined in mice by 10 mg/kg (ip).
3.6. Controlling Multiple Parameters
In fact, recent works adjusting brain exposure did not always aim at one physicochemical property but used a combination of various strategies. Occasionally, the change in a single parameter cannot meet the expected requirements, or it would cause the deterioration of other important characteristics, such as activity and pharmacokinetic parameters. Investigators controlled multiple parameters to balance BBB penetration and other properties.
Chen et al. sought to optimize compound 42 (LY2881835), a free fatty acid receptor 1 [FFAR1, also known as G protein-coupled receptor 40 (GPR40)] agonist with concerns about CNS penetration. GPR40 is an excellent target for type 2 diabetes mellitus because it regulates glucose homeostasis by two mechanisms, glucose-stimulated insulin secretion and incretin secretion. However, GPR40 might also be connected with antinociception, adult neurogenesis, and neurovascular degeneration. (109) Compound 42 had a certain amount of CNS exposure (Cbrain/Cplasma = 0.14). In addition, 42 displayed a high clearance in vitro (t1/2 = 18 min in human liver microsomes, and t1/2 = 2 min in mouse liver microsomes) and a short half-life in vivo (t1/2 = 1.3 h). After exploration at multiple structural sites, the study discovered that carbonyl replacement increased tPSA and decreased LogP. Then, cleavage of the highly rigid spiro[indene-1,4′-piperidine] ring enhanced molecular flexibility. Introduction of a nitrogen atom into the piperidyl group further restricted the entry of molecules into the brain. Compounds 43 and 44, without concerns about CNS penetration, possessed prolonged half-lives and exhibited good agonistic activities together (Figure 17). (110)
Figure 17
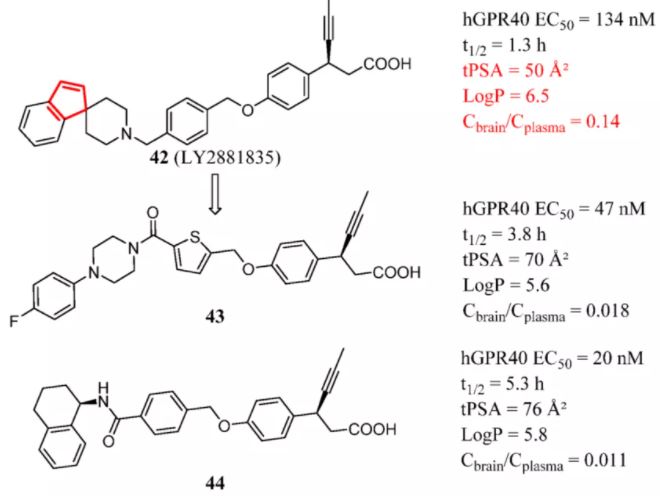
Figure 17. GPR40 agonists: controlling multiple parameters to reduce BBB penetration. Cbrain/Cplasma of 42 was determined in mice at 1 h after 30 mg/kg (po). Cbrain/Cplasma of 43 and 44 was determined in mice at 0.75 h after 30 mg/kg (po).
A simple structure makes it easier to pass through the BBB. Abandoning part of the structure of the compound led to a smaller MW, less HBD, and a lower tPSA. However, it may affect target activity. Medicinal chemists made extensive attempts to remove the fragments that make the smallest contribution to binding. Synthetic intermediates have the potential for further modification. As disclosed by Chen et al., compound 45 was a μ-opioid receptor (MOR)/κ-opioid receptor (KOR) agonist. (111) In particular, compound 45 is not a morphine-like structure, providing an opportunity to develop a small molecule opioid receptor agonist unaccompanied by adverse effects. One of the synthetic intermediates of compound 45 exhibited weak MOR agonism activity and was determined to be a lead compound to develop a series of N-(1,2,3,4-tetrahydro-1-isoquinolinylmethyl) benzamides with MOR agonism. Finally, paradimethylbenzylic acid amide (46) and m,p-dichlorobenzylic acid amide (47), both bearing methyl amine analogues, were synthesized. They exhibited potent activity with EC50 values of 10 nM. In addition, the brain concentrations of analogues 46 and 47 were approximately 5- and 8-fold greater, respectively, than that of 45 (7424.0 and 11696.0 ng/g, respectively). This confirmed that simplifying the structure facilitates the CNS distribution (Figure 18). (111)
Figure 18
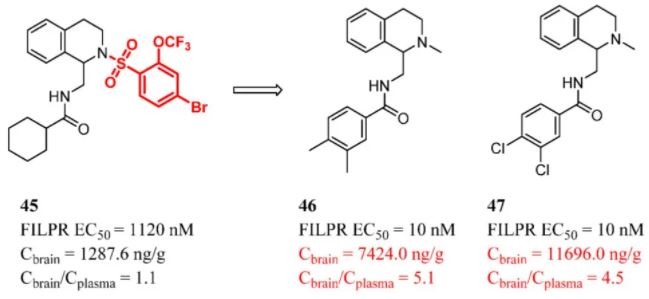
Figure 18. Opioid receptor agonists: simplifying structure to improve BBB penetration. Cbrain and Cbrain/Cplasma were determined in mice at 0.8 h after 3 mg/kg (iv).
Therefore, in the discovery of CNS drugs, investigators should pay attention to the introduction of extra fragments, especially groups possessing heteroatoms, HBDs, basic, or large MWs. These fragments may adversely affect brain exposure. For example, McGowan et al. tried to optimize the pharmacokinetics of muscarinic acetylcholine receptor subtype 5 (M5)-negative allosteric modulators (NAMs) and maintained high selectivity and high CNS penetration. The chlorine atom was installed by the aniline moieties. The series displayed a very short elimination half-life (t1/2 < 30 min). Moreover, unenhanced activity and no detectable brain levels of compound 49 were unacceptable. This might be attributed to the introduction of cyclobutyl(methyl)amino group, which increased the MW, tPSA, and pKa together. After the previous strategy of modification had been abandoned, incorporation of a single 3-methyl group into the phenol ring acting as a metabolic shield provided the desired short half-life (t1/2 = 2.3 h) in rats. It decreased rat t1/2 by ∼35-fold while maintaining M5 NAM potency and favorable CNS penetration (Figure 19). (112)
Figure 19
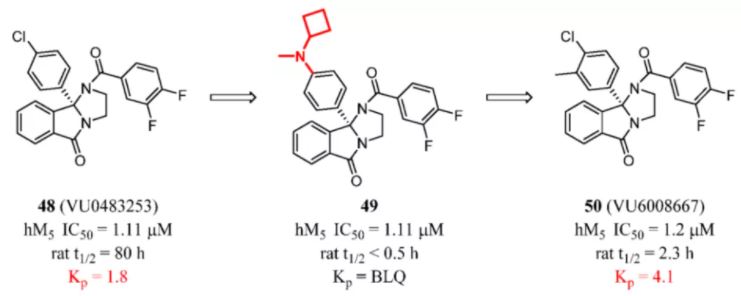
Figure 19. M5 inhibitors: complex structure leads to low brain exposure. Kp was determined in rat by 3 mg/kg (po). BLQ indicates below the limit of quantitation.
4. Hijacking Carrier-Mediated Transcytosis
Among transportation routes, solute carrier (SLC) superfamily proteins, as membrane transporters, are involved in conveying various small molecules. (113) Some essential nutrients such as glucose and amino acids require CMT to reach the brain. They provide a feasible opportunity for drugs with a low level of diffusion to cross the BBB. Connecting fragments that can be recognized by specific transporters strongly expressed at the BBB significantly improves the cellular and brain uptake of drugs. Moreover, because prodrugs effectively decrease the concentration of dissociative parent drugs in circulation after being attached to this shuttle, off-target effects simultaneously decrease or even disappear. (114)
Hijacking CMT is usually achieved by designing prodrugs. The prodrug needs to be recognized by transporters and transported across the BBB. Then the active molecules are released by efficient enzymatic bioconversion. (115) Therefore, an effective prodrug that can deliver drugs to the brain should possess the following characteristics. (1) The prodrugs should possess sufficient stability in peripheral circulation so that the drug can survive before entering the brain. (2) The structure has to be recognized by CMT protein and can consequently compete with endogenous nutrients. (116) (3) The prodrug should be specifically hydrolyzed to release the parent drug at the target site. (117) In addition, safety must also be taken into consideration. The most striking characteristic of CMT-utilizing compounds is competition with nutrients. Therefore, such structural design strategies should pay attention to the fact that the prodrug should not affect the requirement of essential nutrients in the brain. (118)
l-Type amino acid transporter (LAT1)-mediated prodrugs are the most common design utilizing LAT1 for brain delivery. LAT1 is a transmembrane heterodimeric protein that is selectively and strongly expressed at the luminal and abluminal sides of the BBB. Because the brain requires a constant supply of amino acids, LAT1 is strongly expressed in neurons, astrocytes, and microglia. (119) Several well-known clinically used amino acid-mimetic drugs and prodrugs are transported by LAT1, such as L-dopa, gabapentin, and melphalan. (120) Moreover, LAT1 utilization does not interrupt brain amino acid homeostasis, and its expression and function are not altered by inflammatory insult. (121)
In recent years, many studies have designed LAT1-mediated prodrugs. Puris and colleagues designed five newly synthesized LAT1-utilizing prodrugs of the cyclooxygenase inhibitor ketoprofen. After a single dose of 25 μmol/kg (ip) in mice, prodrugs 51 and 52 were able to release reasonable concentrations of the parent drug in the brain. Importantly, the brain distribution for the unbound parent drug released from prodrugs 51 (Kp,uu = 0.13) and 52 (Kp,uu = 0.35) was >10 times higher than that for ketoprofen itself (Kp, uu = 0.01) (Figure 20). (122)
Figure 20
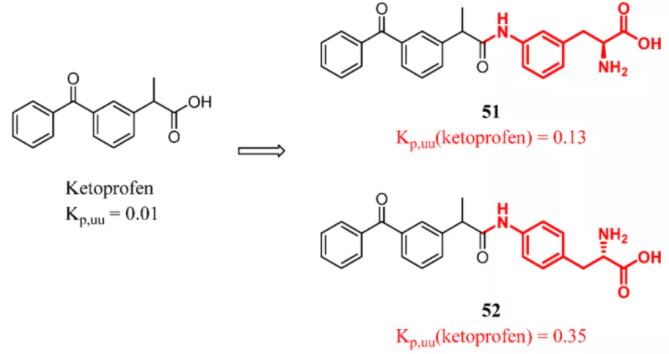
Figure 20. Ketoprofen: introducing a constitutional unit recognized by LAT1. Kp,uu was determined in mice by 30 mg/kg (po). Kp,uu(ketoprofen) represents the Kp,uu of ketoprofen released by compounds 51 and 52.
Dhanawat et al. designed and synthesized a LAT1-connected nipecotic acid prodrug. Using carrier-mediated transport, the nipecotic acid prodrug improved penetration through the BBB and showed antiepileptic activity due to the potent inhibition of neuronal and glial-aminobutyric acid (GABA) uptake. Thirty minutes after treatment with nipecotic acid and the nipecotic acid prodrug (15 mg/kg), no nipecotic acid was detected in the brains of mice in nipecotic acid group, while the concentration of nipecotic acid released by the prodrug group was 450 nmol/g. The nipecotic acid prodrug greatly enhanced the brain transport of nipecotic acid, justifying the rationale of design (Figure 21). (123)
Figure 21
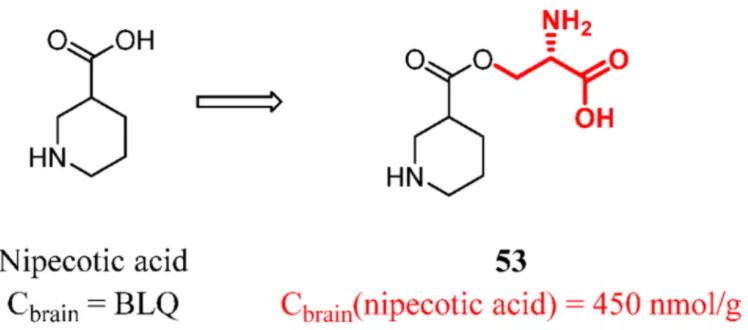
Figure 21. Nipecotic acid: introducing a constitutional unit recognized by LAT1. Cbrain was determined in mice at 30 min after 15 mg/kg (iv). BLQ denotes below the limit of quantitation. Cbrain(nipecotic acid) represents the Cbrain of nipecotic acid released by compound 53.
Montaser et al. developed four novel LAT1-utilizing prodrugs of four nonsteroidal anti-inflammatory drugs. The small-sized LAT1-utilizing prodrug of salicylic acid successfully delivered the parent drug across the BBB. The salicylic acid prodrug was able to cross the BBB at a rate 5 times higher than that of salicylic acid itself, and it released its parent drug specifically in the mouse brain (Figure 22). (124)
Figure 22

Figure 22. Salicylic acid: introducing a constitutional unit recognized by LAT1. Kp was determined in mice by 25 μmol/kg (po). Kp(salicylic acid) represents the Kp of salicylic acid released by compound 54.
l-Ascorbic acid (AA) was also reported as a carrier promoting brain drug delivery. There are two transport proteins in the brain that can transport AA. Glucose transporter 1 (GLUT1) can transport the oxidized form of AA, which is then reduced into AA and will be retained in the brain and enhanced to its CNS level, while Na+-dependent vitamin C transporter (SVCT2) transports AA directly into the brain. (125) Qiu et al. recently combined AA and organic amine as carriers for ibuprofen delivery. Novel prodrug 55 exhibited excellent ability to cross the BBB. The relative uptake efficiency was enhanced to 3.16 times that of naked ibuprofen (Figure 23). (126)
Figure 23
.JPG)
Figure 23. Ibuprofen: introducing a constitutional unit recognized by AA. Cmax,brain and AUCbrain were determined in mice by 48 mmol/kg (iv). The Cmax,brain of ibuprofen was observed 25 min after administration. The Cmax,brain(ibuprofen) of compound 55 was observed 60 min after administration. Cmax,brain(ibuprofen) represents the Cmax,brain of ibuprofen released by compound 55. AUCbrain(ibuprofen) represents the AUCbrain of ibuprofen released by compound 55.
Hijacking CMT, a prodrug can successfully increase brain exposure. However, sometimes the concentration of the parent drug released by the prodrug in the brain does not increase with the prodrug. The prodrug is unstable and decomposes in the peripheral circulation or cannot be hydrolyzed into the parent drug in the brain. Puris et al. connected ferulic acid and amino acid residues. Amide-based prodrugs with an aromatic ring in the promoiety were effectively bound to LAT1 and crossed the BBB. However, the brain exposure level of ferulic acid released by prodrugs was not significantly improved. This result indicated that compound 56 is difficult to hydrolyze into the parent drug (Figure 24). (127)
Figure 24
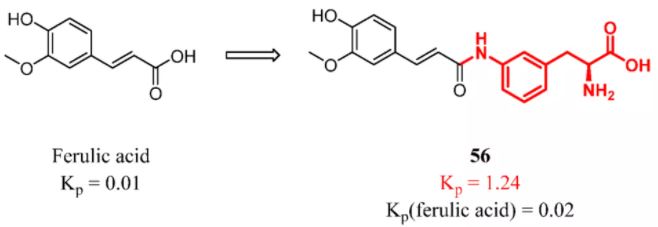
Figure 24. Ferulic acid: introducing a constitutional unit recognized by LAT1. Kp was determined in mice by 25 μmol/kg (po). Kp(ferulic acid) represents the Kp of ferulic acid released by compound 56.
Therefore, there is another design idea that is different from the prodrug. The delivery is bound to the molecule without affecting the activity of the target. The advantage of this strategy is that there is no need to consider the stability and hydrolysis of the prodrug. For example, Messeguer and co-workers synthesized a library of CR-6 derivatives linked to endogen nutrients, among which amino acid compound 57 had the highest brain exposure. The structural modification of the CR-6 scaffold did not impair the protective action exerted by the library members. Prodrug 57 and parent drug CR-6 were ip administered in rats at a dose of 0.2 mmol/kg. After 1 h, Cbrain in the prodrug group was 15.8 nmol/g, approximately 4-fold more than in rats treated with CR-6 (Figure 25). (128)
Figure 25
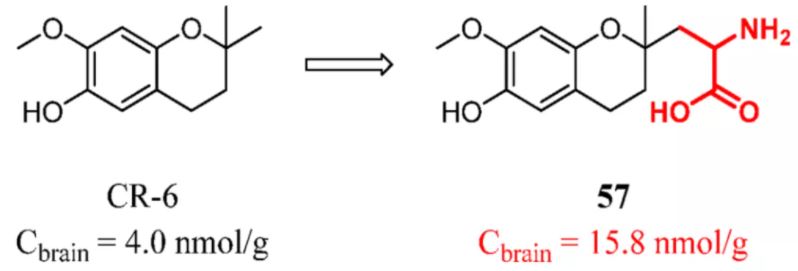
Figure 25. Potent reactive oxygen species (ROS) and reactive nitrogen species (RNS) scavenger agent CR-6: introducing a constitutional unit recognized by LAT1. Cbrain was determined in rats at 1 h after 0.2 mmol/kg (po).
5. Special Structural Targeting of CNS
There are some very novel structural design strategies that can target drugs into the brain to achieve brain selection. Thus, the targeting is essentially a “locked-in” effect. Specifically, the drug circulates in the blood throughout the body and enters the brain through passive diffusion or active transport. However, the mechanisms of exchange are bidirectional, which means passive diffusion or active transport from the brain to blood proceeds in the meantime. (129) Molecules with special structures can be transformed by the microenvironment in the CNS, such as various enzymes, and then cannot be transported outside across the BBB once they enter the CNS. With a “lock-in” function, the transportation of prodrugs is changed from bidirectional to unidirectional, which enhances the concentration of active drugs in the brain. (130)
Levacher and colleagues recently reported donepezil-based “biooxidizable” prodrugs. A 1,4-dihydropyridine ring was introduced to replace the piperidine moiety of donepezil. This prodrug approach rationally shielded the positive charge at the nitrogen. Therefore, the 1,4-dihydropyridine nitrogen of prodrug 58, which was not basic enough to be protonated at physiological pH, could cross the BBB because of its good lipophilicity. Once the prodrug is in the brain, a redox activation step mediated by oxidoreductases converts 1,4-dihydropyridine prodrug 58 into the corresponding pyridinium drug 59. The presence of a permanent positive charge within pyridinium drug 59 was expected to entail a “locked-in” effect in the brain. Prodrug 58 had good stability in human plasma and was effectively converted back to the parent drug under various mildly oxidizing conditions (Figure 26). (131) However, during the development of CNS drugs adopting this strategy, toxicity and safety should be mostly taken into account, because there is a similar mechanism with the formation of neurotoxic 1-methyl-4-phenylpyridinium (MPP+). MPP+ is generated from oxidized 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in the brain, which has been implicated as a cause of PD. (132) Levacher and colleagues accomplished a series of toxicological experiments. Compound 59 did not show genotoxicity in vitro and displayed high LD50 values in mice. In addition, compound 59 did not accumulate in the brain after repeated daily administration. (131)
Figure 26
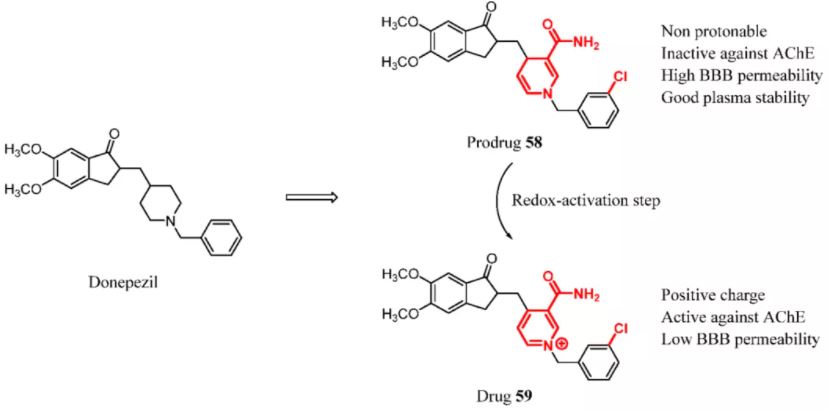
Figure 26. Donepezil-based “bio-oxidizable” prodrugs.
Wu et al. developed a brain targeting AA prodrug with “lock-in” function. The ibuprofen prodrug was delivered by an AA carrier and was combined with a thiamine disulfide delivery system. Prodrug 60 was stable and convenient for conservation in air. Once it entered the CNS, it could be reduced by disulfide reductase and then ring-closed to be a thiazolium that “locked in” the brain. The thiazolium cannot be transported outside across the BBB. Then the prodrug system can release the active drug by hydrolysis to play a therapeutic role. The prodrug exhibited excellent ability to be transported across the BBB in the biodistribution experiment (Figure 27). (133)
Figure 27
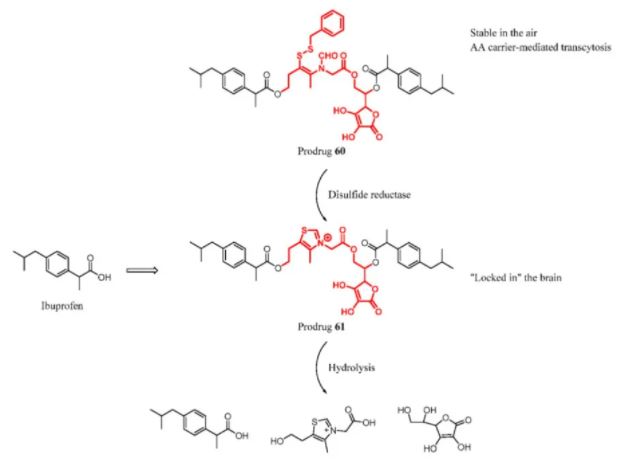
Figure 27. Ibuprofen prodrug with a thiamine disulfide delivery system.
6. Conclusion and Outlook
In conclusion, with the continuous deepening of research on the CNS, it is encouraging that CNS diseases will be overcome. The main challenge faced by medicinal chemists is to seize opportunities amidst the continuous emergence of new discoveries.
https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00910
Copyright© Shanghai VastPro Technology Development Co., Ltd. 2016-2019 Supported by ChinaChemNet Toocle Copyright Notice
4th Floor, Building 9B, 100 Haike Road, Pudong New Area District, Shanghai, 201210 P. R, China.
Tel: +86-21-20608178 Fax: +86-21-20608171 Contact us


